Open Access
Case Report
Max Screen >>
ISSN: 2456-5482
Copyright: © 2016 Crews J. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
A 14 year-old previously healthy Hispanic female was noted to have pancytopenia at an emergency center visit where she was evaluated due to 2 weeks of cough, malaise, and low-grade fever. She was prescribed Azithromycin and Methylprednisolone, but never began the steroid. The pancytopenia persisted over four weeks, with subsequent finding of Coombs-positive warm autoantibodies. The combination of immune thrombocytopenia and immune hemolytic anemia classified the patient with Evans Syndrome. With further monitoring, the hemolytic anemia remained stable and therefore no medical treatment was initiated. Her laboratory findings were presumed due to her recent respiratory infection and began to spontaneously resolve over the next several weeks. This paper discusses the interesting presentation of Evans Syndrome with pancytopenia as well as the differential diagnosis for Evans Syndrome.
Keywords: Evans Syndrome; Pancytopenia
List of abbreviations: CBC: Complete blood count; CMP: Complete metabolic panel; CRP: C-reactive protein; LDH: Lactate dehydrogenase
A fourteen-year-old previously healthy female was found to have persistent pancytopenia following an acute upper respiratory illness, diagnosed as bronchitis at a local emergency center. On further testing with Hematology/Oncology, she was found to have Coombs-positive warm autoantibodies in her serum with hemolytic anemia in addition to thrombocytopenia. This combination of symptoms classified her with Evans Syndrome. This presentation of Evans Syndrome with autoimmune neutropenia agrees with a few studies in the literature. Evans Syndrome is a rare hematologic disorder characterized by Coombs-positive hemolytic anemia and immune thrombocytopenia. The incidence is unknown and it typically affects the pediatric population but with no known gender predilection. The diagnosis of Evans Syndrome is essentially a diagnosis of exclusion and should be considered in a patient presenting with autoimmune hemolytic anemia, immune thrombocytopenia, or both.
A previously healthy fourteen-year-old Hispanic female presented to the general pediatrics clinic for follow-up regarding a visit to an emergency department the day prior. This patient was known to the pediatric clinic prior to the visit. The patient had no significant medical history except obesity, no prior hospitalizations or surgeries, and immunizations were up-to-date. She had no dietary restrictions and family history was remarkable only for her mother having anemia due to heavy menstrual bleeding which subsided following a hysterectomy.
Her parents took her to the emergency department because of two weeks of productive cough, generalized weakness, malaise, and low grade fever (maximum temperature 100 Fahrenheit). Additionally, she had decreased appetite for one month. The emergency department diagnosed her with bronchitis and discharged her with a five-day dose-pack of Azithromycin and a six-day dose-pack of Methylprednisolone. At that visit, she was noted to be pancytopenic (Figure 1). Mom did recount bruising on the patient’s left shoulder and left popliteal region two weeks prior but the patient believed she had trauma to those areas. Review of symptoms was otherwise unremarkable. The patient noted symptomatic improvement over the previous few days. On physical exam, the patient had vital signs within normal limits for her age. With the exception of acanthosis nigricans on the posterior neck folds and hepatomegaly to three centimeters below the costal margin, physical exam was unremarkable (Table 1).
Repeat laboratory studies showed persistent pancytopenia with no elevation in reticulocyte count. Viral studies including parvovirus, Epstein-Barr virus, and Hepatitis A, B, and C were negative. Patient had repeat laboratory studies three days later showing persistent pancytopenia with decreasing platelet count. The patient was referred to the Hematology/Oncology service for further evaluation.
Two days later, a bone marrow biopsy was performed which showed hypercellularity but no evidence of an infiltrative process. After several days, the patient was noted to be Coombs positive with warm autoantibodies present. The constellation of pancytopenia, positive Coombs test, and hypercellularity on bone marrow evaluation (suggestive of immune-mediated peripheral destruction) defined her diagnosis of Evans Syndrome. Further follow-up laboratory studies showed worsening thrombocytopenia and neutropenia but patient remained asymptomatic from a bleeding and infectious standpoint. Additionally, her anemia was stable which would have served as the triggering factor for treatment initiation. She was therefore monitored without medical intervention.
After multiple laboratory evaluations showing persistent pancytopenia with waxing and waning platelet counts, the patient remained asymptomatic and had continued follow-up with the Hematology/Oncology service. At each visit, it was noted that the patient was nervous discussing possible diagnoses and further interventions. Given that the patient’s anemia was stable with no worsening, no medical intervention was initiated.
Throughout these many interactions with the family, only English was used. The family’s predominant language at home was Spanish, but the family insisted on participating in visits in English because they were essentially fluent in the language, despite being offered teleconferenced and live interpreter use. This did not appear to affect history gathering nor patient care at any level and the family was receptive to continuing medical care in English rather than enlisting the use of an interpreter service.
Evans Syndrome is a rare hematologic disturbance comprised of immune thrombocytopenia and Coombs-positive immune hemolytic anemia [1]. The incidence is unknown but it is thought to be a rare disorder, mainly affecting the pediatric population but with no known gender predilection [2]. This diagnosis is essentially one of exclusion and should be considered in a patient presenting with autoimmune hemolytic anemia, immune thrombocytopenia, or both. Diagnosticians should bear in mind that one laboratory finding may precede the other [3]. The specific antibodies that cause thrombocytopenia and hemolytic anemia are unknown, but current research suggests the combination is more likely profound immune dysregulation rather than a coincidental combination of cytopenias [1]. There are studies demonstrating approximately half the cases of Evans Syndrome are associated with other conditions, including infections (Hepatitis C, HIV), lupus, and lymphoproliferative disorders [1]. Findings of bi-or pancytopenia (as noted in this patient) should prompt health care providers to search for an underlying diagnosis [4]. The percentage of pediatric patients with concomitant neutropenia and Evans Syndrome (e.g. pancytopenia) is unknown, but a case series of adults showed only 14.7% of patients had concurrent autoimmune neutropenia (defined as neutrophil count less than 1.5 x 109/L on 2 occasions separated by at least 1 week) at the time of diagnosis of Evans Syndrome [1].
The etiology of pancytopenia in children varies widely, ranging from transient viral marrow suppression to malignant bone marrow infiltration. When a patient presents with pancytopenia, work-up should include a complete blood count including red blood cell indices (i.e. MCV and MCH), reticulocyte percentage, and examination of the peripheral blood smear.
Evaluation for hemolysis includes indirect bilirubin, lactate dehydrogenase, and haptoglobin. Consider direct Coombs testing; the diagnosis of autoimmune hemolytic anemia is made with the combination of hemolytic anemia (anemia, elevated lactate dehydrogenase, low haptoglobin, high indirect bilirubin), spherocytes on peripheral blood smear, and positive direct Coombs test. Performance of bone marrow biopsy should be considered heavily, in conversation with a hematologist/oncologist.
This case serves as a reminder of the broad differential diagnosis for a patient with pancytopenia. Congenitally acquired causes for cytopenias include Fanconi Anemia, dyskeratosis congenita (supported by nail dystrophy and pulmonary fibrosis), ShwachmanDiamond Syndrome (pancytopenia with pancreatic insufficiency) and congenital amegakaryocytic thrombocytopenia (typically with history of severe thrombocytopenia as a neonate) [5]. Acquired aplastic anemia has many causes, including medications (chloramphenicol, nonsteroidal anti-inflammatories, anti-thyroid drugs, corticosteroids, penicillamine, allopurinol, and gold) and infections. Classically Parvovirus B19 causes megaloblastic anemia but several case studies have shown aplastic anemia. Additionally, Epstein-Barr virus, HIV, and hepatitis may cause aplastic anemia [5]. Autoimmune diseases may initially present with pancytopenia, including systemic lupus erythematosus. Nutritional deficiencies may cause pancytopenia including copper, folate, and B12 deficiency. Breastfed infants of Vitamin B12-deficient mothers are also at risk for deficiency [6]. Vitamin-D dependent rickets in breastfed infants has reportedly caused bone marrow fibrosis and pancytopenia. Also on the differential are processes that infiltrate the marrow space, such as leukemia and lymphoma [7,8]. Finally, providers should consider genetic syndromes including DiGeorge Syndrome and cartilage-hair hypoplasia (absolute lymphocyte counts and T-cell counts for age are generally low, with neutropenia reported) [9].
The patient discussed in this case had cytopenias revealed during a workup for her viral-type symptoms, a classic clinical progression. One limitation of this case is the lack of medical intervention which is often used in Evans Syndrome, including glucocorticoids, IV immunoglobulin, Cyclosporin A or immunomodulatory agents such as Mycophenolic acid [1,3,4]. Notably, the few existing studies regarding treatment have shown high rates of treatment resistance or failure [3].
Evans Syndrome, though a relatively rare diagnosis, should be considered when presented with a patient with pancytopenia. The differential is broad but keeping this diagnosis in mind could drastically change the course of management.

![]()

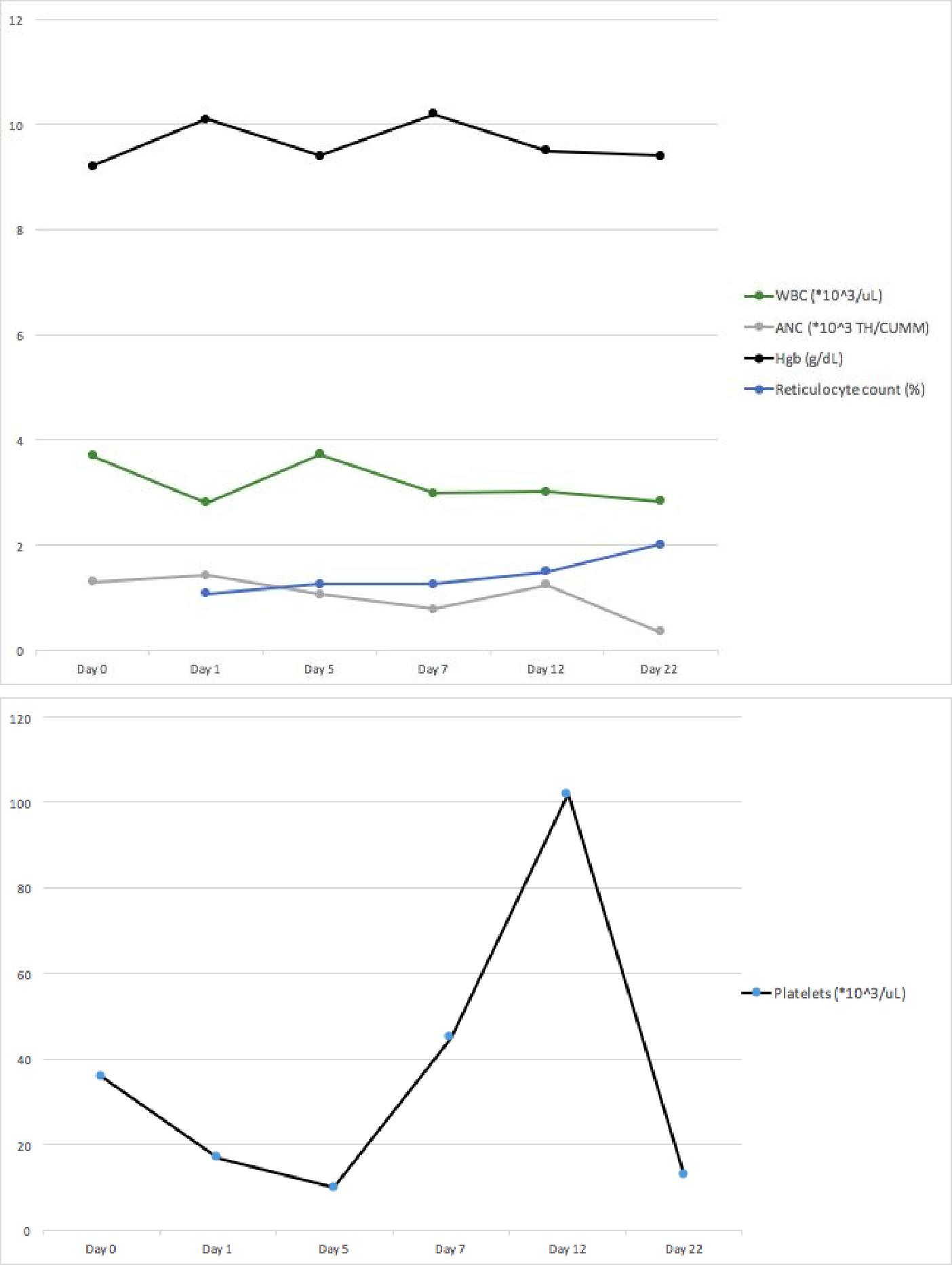 |
Figure 1: Patient’s laboratory data over time |
| Day 0 | Initial emergency center visit. Diagnosed with bronchitis, prescribed Azithromycin for 5 days and Methylprednisolone for 6 days |
Day 1 |
General pediatrics clinic for emergency center follow-up due to abnormal laboratory studies. Differential at that time included leukemia/lymphoma (pancytopenia, hepatomegaly, questionable recent bruising), hepatitis with viral prodrome, Epstein-Barr Virus infection (malaise, fever, cough). Instructed to continue Azithromycin and stop steroids. Ordered labs including CBC, reticulocyte count, CRP, CMP, monospot, uric acid, LDH, parvovirus titer, Hepatitis panel, and chest x-ray. At that time, pancytopenia persisted with no elevation in reticulocyte count. CMP unremarkable, with no transaminitis. Monospot was negative and other labs pending. |
Day 5 |
General pediatrics clinic follow-up for laboratory results. Chest x-ray without pneumonia or mediastinal mass. Pancytopenia persists with decreasing platelet count. Leading diagnosis was leukemia/lymphoma versus viral suppression of bone marrow. |
Day 7 |
Hematology/Oncology appointment. Bone marrow biopsy performed |
Day 9 |
Phone message -Bone marrow pathology showed no leukemia. Mother confronted patient about skipping meals at school; patient admitted to 2 months of eating only one light meal per day to avoid having her body "damaged" due to obesity |
Day 12 |
Hematology/Oncology follow-up appointment for bone marrow biopsy results. Constellation of newo-nset pancytopenia, positive Coombs test, hypercellularity on bone marrow biopsy suggestive of an immune-mediated peripheral destructive process (warm autoimmune hemolytic anemia). By definition, diagnosed with Evans Syndrome. Differential at this point also included other autoimmune and immune dysregulation phenomena such as lupus, rheumatoid arthritis, autoimmune lymphoproliferative syndrome, or chronic variable immunodeficiency. Labs sent for anti-neutrophil antibody, antiplatelet antibody, Coombs test. Cytopenias beginning to spontaneously improve, so may have been infection-triggered autoimmune event, but possible to have persistent immune-mediated cytopenias. |
Day 22 |
Hematology/Oncology appointment. Thrombocytopenia worsening but no bleeding symptoms. Neutropenia worsening. Due to the autoimmune hemolytic anemia not worsening, treatment withheld [(would be immunomodulatory agents like CellCept (Mycophenolic acid)] |
Day 26 |
General pediatrics follow-up appointment. Discussed diet, multivitamins and iron replacement with trace minerals supplementation. |
Table 1: Timeline of Events |
|




































