Open Access
research article
Max Screen >>
ISSN: 2576-7690
Copyright: © 2014 Sabharwal NS. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Mutation in a single nucleotide of a gene has the potential to change the structure and/or function of its protein. Albeit simply saying, it is not observed to be a general phenomenon. The effect of mutation is primarily determined by the stereochemical nature of the amino acid which has replaced the previous amino acid, resulting in the residue location being affected. Here we show that despite a change in the frequency of occurrence of a particular amino acid in a particular protein in different types of organisms, the overall function of the protein can still remain unaffected, even when the resultant protein conformation is relatively altered. Phylogenetic trees were constructed for the proteins belonging to the same family on the basis of the sequences extracted from protein structures. Variation in the percentage of every existing amino acid of each of the considered protein is further calculated. In contrast to this sequence based mutual comparison of proteins, structural comparison is also computed in terms of standard TM_Score and alteration in the count of structurally similar residues falling within the 5Å distance deviation. The functional and structural role of an evolutionary alteration or mutation in a protein sequence and its concomitant effect on the protein structure is thus analyzed.
Keywords: Villin; Headpiece; Sub-domain; Evolution; Topology
Villin is one of the major cytoskeleton proteins that bind to actin. It is localized to some specific tissues like the microvilli of intestine and kidney [1]. Some of the cell types present in the unorganized brush border in the pancreatic and bile duct also have villin, although at very low concentration. These cells help in the absorption process exactly similar to functional microvilli of intestine and kidney. Villin is also expressed in intestinal cells of the embryo [2]. Villin belongs to a large class of actin regulating proteins. Regulation of the actin filaments is performed by actin binding proteins. These proteins assist the sequestration of actin monomers, severing and cross-linking of filaments, and cover their ends [3]. In humans, the villin is encoded by two mRNAs of dissimilar lengths and surprisingly, the coding region of their cDNAs shows no difference.
In culture conditions, the mRNA expression level is also found to be similar in differentiated intestinal cells and the villin expressing microvillus tissues [4]. Here begins the interesting aim of our study to depict the amino acids which show evolutionary changes or evolve more often in a protein. We also screen the amino acids which normally remain conserved in a protein and thus define its peculiar conformation. We have studied the effect of evolutionary alterations in the frequency of occurrence of amino acids on its respective protein structure. For computational ease and small size, we have selected the Villin Headpiece as our model protein. Furthermore, as it is naturally available in most of the organisms, the application of this study can be extended to a wide range of organisms.
Villin protein structure consists of seven domains, six of which are present on the N-terminal and one domain is present on the C-terminal. Six N-terminal domains make up the N- terminal core and the C-terminal domain constitutes the villin Headpiece [5,6].
A short fragment of amino acid residues is repeated six times in the N-terminal core, with each domain sharing a repeat [7].These structurally similar repeats are present in all actin-severing proteins viz. gelsolin, fragmin and severin [7]. The Headpiece engulfing the C-terminal domain does not show significant sequence similarity with other regions of the villin [7]. Both C and N-terminal core engulf actin and calcium binding sites [5,6]. The Headpiece is believed to be evolved by a recombination event between gelsolin and some other actin binding protein genes. These genes are believed to be synapsin-I (an acting bundling protein present in the synaptic vesicles), as it shares significant homology with the Headpiece [8]. Villin structure and function is related to two groups of proteins, one consisting of a bundling protein Quail (found in drosophila) and protovillin (found in Dictyostelium), and the other group including gelsolin and scinderin proteins found in higher eukaryotes [9,10], whereas fragmin and severin proteins are normally found in lower eukaryotes [11,12]. The first group of proteins encodes seven domains, as in the case of villin, and the second group represents the domain arrangement exactly similar to villin core [13].
The 76 residue long villin Headpiece (HP) is also present in many other actin bundling proteins [14,15]. It is furthermore intriguing to observe that removal of first 9 residues from C-terminal domain results in the construct, normally represented as HP-67, and this partial truncation does not affect the overall function and stability of the HP Protein [15,16]. Furthermore, this HP-67 structure consists of two Sub-Domains (SD), one present at the N-terminal and the other one localized at the C-terminal. The latter 35 residue SD consists of 3 helices forming a hydrophobic core and is generally represented as HP-35 [16,17]. This HP-35 is one of the shortest, naturally occurring proteins to show cooperative and rapid folding probably due to its relatively short size engulfing high helical content [16,18,19]. Also quite noteworthy, three phenylalanine residues partly stabilize this conformation [20] and the evolutionary conserved TRP64 additionally plays a major role in its interaction with F-actin [21].
It is also well known that the villin protein is associated with actin bundling, severing, nucleation and capping, which regulate the actin filaments. Villin due to N-terminal half of its core, is found to severe actin filaments under high calcium concentrations [22]. Such actin binding sites are present on both N-terminal and C-terminal, the former being regulated in calcium dependent manner while the latter one shows a calcium independent activity. When the calcium concentration is more than 10-4 M, villin severing activity produces short filaments by acting upon F-actin.
Conversely, at lower concentration range of 10-7 to 10-6 M, villin prevents elongation of actin filaments by capping whereas at substantially lower concentration (lesser than 10-7 M) it shows the bundling activity [23]. Unlike the severing activity, bundling activity of villin is normally balanced by other in-vivo proteins [23]. Similarly, phosphorylation of tyrosine residue in villin results in its decreased affinity for F- actin, decrease in nucleation activity and substantial increase in its severing action [24]. There is yet another interesting aspect of villin structure. It is cleaved by trypsin into two fragments, one encompassing domain1 to domain3 and the other one encoding domain4 to domain7. These fragments inclusive of the referred SDs are respectively known as 44T and 51T [7]. Here the presence of calcium is very important. In presence of calcium, the proteolytic cleavage between domain2 and domain3 is inhibited and such cleavage stays unchallenged in the presence of EGTA (Ethylene Glycol Tetra Acetic acid). It could be probably due to alteration in the N-terminal segment conformation in the presence of calcium [7].
Protein structures related to villin HP and SD were downloaded from the Protein Data Bank (PDB). A total of 32 PDB files were downloaded and their structural records were converted to FASTA format through an in-house PERL script. These structures were then divided into three groups according to their encoded count of amino acids. These structures along with their FASTA sequences with around 67 residues were categorized as "villin Headpiece" (Group HP) (1QQVA, 1QZPA, 1UJSA, 1YU5X, 1YU7X, 1YU8X, 1ZV6A, 2K6MS, 2K6NA, 2RJVA, 2RJWA, 2RJXA, 2RJYA, 3MYAA, 3MYCA, 3MYEX and 3NKJA). Similarly the structure and sequence files with around 35 residues were classified as "villin Sub-domain" (Group SD) (1UNCA, 1UNDA, 1VIIA, 1WY3A, 1WY4A, 1YRFA, 1YRIA, 2JM0A, 2PPZA, 3IURA, 3TJWA, 3TRVA, 3TRWA and 3TRYA). The lastly considered group comprised of substantially bigger villin structure (3FG7) with 398 amino acids, which was thus excluded from our sequence and structural comparison with other considered structures. Similarly, 3IURA was also removed from the SD group, as it encodes three chains A, B and C with 684, 6 and 5 amino acids respectively and that is nowhere comparable to the default chosen set size of 34-36 amino acids encoded in all the aforementioned SD structures. So finally, a total count of 30 structures (17 for HP and 13 for SD) were considered for further analysis.
Structural FASTA information extracted from the selected HP and SD structures was employed to construct 3 phylogeny trees (One each for HP, SD and one for both of them) using an online tool. For its custom support and ease of usage, we preferably employed this online server and it made the analysis quite handy. These phylogeny trees are well illustrated in Figures 2, 3 and 4 respectively. Through these trees, mutual evolutionary distances among the selected structures were manually computed, as enlisted in tables 1, 3 and 4 respectively.
Here, we computed the percentage of structurally encoded amino acids in each of the selected protein, through an in-house PERL script with the consideration of modified amino acid entries (Heteroatom) as the normal residues. The heteroatom consideration allowed us to unanimously exploit and extrapolate specific amino acid position and availability for all the considered evolutionarily linked protein structures. It further resulted in data showing the percentage of every amino acid encoded in HP and SD structures, as represented in Table 5 and 6.
It also allowed us to screen the evolutionarily selected and variant amino acids (represented as boldface in the tables), with the arbitrarily selected threshold difference of more than 0.2 percent against the respective average data. To compare the percentage of every encoded amino acid and the structural similarity of compared proteins, we further considered the proteins as per their source organism. Through this analysis, we obtained the percentage difference of amino acids across different species, as enlisted in Table 2. To compare our results with the percentage of amino acids generally available in naturally occurring proteins, Table 7 was constructed through the premier information resources [25-27].
Other than the sequence based phylogeny tree distance deviations among the considered proteins for each of the HP and SD group, we also calculated their TM_Score and GDT residues (Global Displacement Test residue within the 5Å distance deviation from the other considered structurally equivalent residue) through TM-Align tool of Zhang's lab. The TM_Score and GDT residue percentage data thus obtained is enlisted in Tables 8 and 9 respectively for HP and SD group proteins.
Considering all the selected proteins encoding Headpiece domain of the Villin structure, we constructed a phylogenetic tree (Figure 2). This snapshot illustrates the evolutionary link between the considered HP structures, and it was used to calculate mutual evolutionary distances amongst the selected structures (Table 1). As per Figure 2, 1QZPA and 1ZV6A are close to each other and the same is referred by their distance value enlisted in Table 1. Contrary to it, 1QZPA and 1UJSA lie far apart in the tree and so their evolutionary distance should be more than that of 1QZPA-1ZV6A, as clearly observed in Table 1. Similarly, the 2K6MS-2K6NA distance is zero and these structures are expected to be evolutionarily very close, which was clearly observed in the tree shown in Figure 2. Such evolutionary distance analysis can be uniformly applied to all the structures (2RJVA to 1YU7X), as shown in Figure 2. Considering the source organism information for these structures (as listed in Table 2), we see that Homo sapiens sequences are mutually closer compared to their correlation against Gallus gallus.
For Headpiece distance analysis, the selected SD structures were also scrutinized through their phylogenetic tree information (Figure 3) which further resulted in their mutual distance matrix, as shown in Table 3. As shown in Figure 3, 1UNDA is closer to 1UNCA than 3TJWA, and it implies that 1UNDA-3TJWA might have evolved much more than 1UNCA-3TJWA, as correctly shown in Table 3.
Similarly 1UNDA, 2JM0A are closely placed in Figure 3, and are thus minimally deviant in Table 3. It also shows that 3TJWA, 3TRYA, 1YRFA, 3TRVA and 1YRIA have evolved almost to same extent and so their mutual distance should be less, as shown in Table 3. Like the aforementioned source organism analysis for HP structures, here also sequences from the same species have showed lower mutual evolutionary distance when correlated with different species.
Besides considering the HP and SD distance analysis individually, we also employed them together for constructing the phylogenetic tree (Figure 4), and its distance data is enlisted as Table 4. Here it is interesting to note that some of the SD entities are evolutionarily closer to the HP structures. Other than the shifted tree localization and the minor mutual distance deviations of these structures, caused due to consideration of more files, the overall outcome is similar as reported.
After all this mutual distance calculation for HP proteins, we calculated the percentage availability of amino acids in the selected structures (Table 5). Here the AVG row represents the average occurrence of a specific residue in all the selected structures and the boldface values are the structural entries with an arbitrary difference of more than 0.2 compared to the average (AVG) value of that amino acid. These boldface entries therefore represent the structures showing a significant difference in the available percentage of an amino acid compared to its average value. The total count of such boldface structures can thus be used to calculate the percentage of amino acid residues (shown as aa% change), that have got significantly altered in the selected HP structures. This analysis can then be plotted to yield Figure 5(a) and 5(b) respectively, representing the average percentage and percentage change for each of the amino acid. This analysis further reveals that leucine is eminently available in each HP structure, and is orderly followed by lysine, glutamic acid, alanine, phenylalanine, aspartic acid, proline, valine, asparagine, arginine, glycine, threonine, serine, glutamine, methionine, tryptophan, tyrosine, histidine, isoleucine and cysteine. Another glance at Figure 5(b) shows one interesting aspect that evolutionary variation amongst the selected structures has preferentially employed certain specific residues viz. valine, threonine, proline, isoleucine, lysine, asparagine, aspartic acid, glutamine, phenylalanine, glycine and arginine, as their available percentage is significantly altered in each of the selected HP structure. Another fold of this analytical story implies that the frequency of occurrence of tryptophan is kept majorly unaltered and the percentage of amino acids is mostly repeated in a similar pattern across different HP structures.
Exactly like the aforementioned HP structural analysis, SD structures were also scrutinized as represented in Table 6 and Figure 6. Here also the boldface values are the ones which have shown a significant change in their percentage, as compared to their average figures. We observed that leucine is eminently available in all SD structures and is orderly followed by lysine, phenylalanine, alanine, glutamine, glycine, glutamic acid or serine, aspartic acid, arginine, asparagine, methionine, proline or threonine, valine, tryptophan, histidine, isoleucine, tyrosine and lastly followed by cysteine.
Quite intriguingly, cysteine is not available in any of the SD structure, be it a natural protein (1VIIA) or any of the other employed synthetic constructs. Furthermore, certain amino acids viz. isoleucine, histidine, lysine, asparagine, aspartic acid, glutamine, tyrosine, methionine, phenylalanine and arginine are plausibly evolutionarily selected for variations, as their availability percentage has been found to be significantly variant across all the selected SD structures. Moreover, two amino acids glutamic acid and serine are not showing variant percentages across the SD structures, and are thus very important for the SD conformation. Same as HP, here also the amino acid percentages are mostly repeated in a similar fashion across different structures.
To cluster the amino acid percentage data, as per the native source organism, we constructed Table 2 which represents that the frequency of a specific amino acid follows an almost similar prototype for a particular source organism. Here we observed that isoleucine is not present in any of the selected non-synthetic or natural proteins with the source organism Gallus gallus, although for Homo sapiens it is present in all the selected structures. Besides this, serine is found in all selected Gallus gallus structures while only in three out of five Homo sapiens proteins. Similarly tyrosine is available in all the Homo sapiens structures, though for Gallus gallus it is found only in three out of thirteen proteins. Likewise, except one Gallus gallus structure, tryptophan is present in all the natural Homo sapiens as well as Gallus gallus proteins. Cysteine is also found to be present only in two Homo sapiens structures, although histidine is found restricted to only a few Gallus gallus and Homo sapiens structures.
After scrutinizing the naturally available structures, a similar brain-storming analysis was performed for the selected synthetic proteins. Here also, isoleucine follows exactly the same trend, i.e. it is unavailable in all the synthetic constructs for the source organism Gallus gallus. Similarly cysteine is unavailable in all the synthetic constructs. Moreover, asparagine, lysine and tryptophan are found in all but one synthetic constructs. Conversely, tyrosine is available only in one synthetic construct. This analysis simply shows that the percentage of amino acids is mostly kept unaltered across synthetically constructed structures also, or in other words, the defined functional attributes are well pertained to the availability and localization of specific amino acids across these structures.
To compare our computed average amino acid percentage figures in our considered structural dataset against the average residue percentage naturally encoded in protein structures, we considered several research works employing a wide array of proteins, ranging from only 118 to 0.55 million (Table 7). Here, AVG-118, AVG-1150 and AVG_0.55_million respectively represents an average amino acid percentage of 118 proteins from different Super-families [25], around 1150 proteins [26] and 5, 49, 616 proteins [27]. Here we have enlisted our computed average amino acid statistics for HP and SD structures as AVG-HP and AVG-SD. For most of the amino acids, we observed an almost equivalent average percentage data among the Doolittle and Carugo referred articles [26,27]. This variation is comparatively higher for Cornish figures [25]. While Cornish and Bowden considered only 118 proteins for the calculation of naturally available amino acid percentages [25], Doolittle increased the sample size to 1150 proteins thereby making the resultant data more reliable. Carugo's work in this regard sounds even more trustworthy, as it considered 549, 616 proteins to compute the amino acid percentages naturally encoded among diverse proteins.
Comparing our observed statistics for HP and SD structures with the aforementioned reference data, we found a substantial variation among residue percentages. This observed difference among the average amino acid percentage of AVG_0.55_million and AVG-HP data is shown as "Substantially Variant for HP" and the similar variation for SD is represented as "Substantially Variant for SD". This observation could be easily explained from the incredibly different data size of proteins considered for both these research works. Secondly, the AVG_0.55_million data encompasses an extremely large set of proteins, including both related and unrelated structures, and our minimal sample size solely comprises of structurally and functionally related proteins.
Although the AVG-HP, AVG_0.55_million amino acid percentage variation is lesser than that of AVG-SD, AVG_0.55_million dataset, the AVG-HP data is significantly different against the AVG_0.55_million information. This is quite predictable also as the structures considered for calculating AVG-HP specifically belong to only villin HP and it becomes fairly obvious to perceive the considerable similarity among the HP proteins. Moreover, according to the research work of Cornish-Bowden "Natural peptides and small proteins in general have amino acid compositions that diverge much more from the average composition of all proteins" [25]. Due to the existing length of our considered SD and HP structures, this Cornish-Bowden statement is valid for our data also, as lucidly enlisted in Table 7.
Subsequent to this sequence analysis of all the selected structures, we computed their structural similarity through TM_Score and GDT residue percentage calculation [28]. As enlisted in Table 8 and for every single structural comparison, here we found a remarkably similar structural topology of the selected proteins in terms of TM_Score. It was also observed that the GDT residue percentage was quite high, which implies a reliable structural similarity of the selected structures. Comparing 1UJSA- 1QZPA and 2K6NA-2K6MS Homo sapiens structures, we found that the former pair showed a TM_Score of 0.61148 in comparison to the latter score of 0.90301 and those protein pairs showed a GDT residue count of 87.17949 and 98.50746 respectively. Hence, we observed that the TM_Score and GDT residue percentage figures individually changed drastically even within the same organism.
Quite interestingly after considering all such protein pairs, we observed that the TM_Score showed a drastic difference in two protein structures even when their GDT residue percentage score was pretty high, being greater than 90% in almost all the cases. Moreover, even when such percentage count of GDT residues was substantially lower, the TM_Score figure followed almost a similar trend. As TM_Score calculation emphasized on the distance deviation in the equivalent residues of the compared protein structures, it nullified their global structural similarity even when they shared a higher count of structurally similar and conserved residues within an allowed distance deviation. Such global structural similarity of proteins can be astutely attributed to the evolutionarily unaltered characteristic core and functional domains.
In a similar way as mentioned above, SD structures were also scrutinized for mutual structural similarity (Table 9). Amongst all the selected SD structures (listed in Table 2), 1VIIA was the only naturally available conformation. Here we observed several TM_score values lesser than 0.5. This implies a high structural dissimilarity and so this workout seems to be a futile structural similarity analysis. Now logically emphasizing, we know that synthetic constructs are developed by altering certain amino acids for studying some specific properties including nucleation and folding kinetics of the considered protein. The GDT residue percentage and TM_Score analysis, as done on HP, is an insignificant and unreliable parameter to structurally study the synthetic construct in reference to the natural proteins. It is because the synthetic constructs encode specific residue alteration(s), which are not ascribed to the natural phenomenon of evolution.
But still comparing these artificial SD structures against the natural 1VIIA, we observed a non-linear difference among GDT residue percentage and TM_Score measures in contrast to the similar amino acid percentage encoded in these proteins. Therefore, it became fairly reasonable to skip them for further analysis. But, we had already employed all the selected SD sequences, mutually sharing similar amino acid percentage along with the HP sequences also following such a trend, for a phylogenetic tree including all the HP and SD structures (as shown in Figure 4). Thus hereafter eliminating the SD files from our consideration, Figure 4 would also become unrealistic for the structural comparison of proteins in our workout. Leaving this very plausible although strange result aside, it is indeed an incredible work that researchers have developed specifically variant conformations, which retain the conserved backbone topology and are encoded for variant functional attributes.
Through mutual structural comparison analysis as per Table 8 information, we screened the highest as well as the lowest TM_Score match, respectively signified as Highest_TM and Lowest_TM in Table 10, to select the most and the least structurally similar structure available for a particular HP protein. The Table 10 also enlists the GDT residue percentage (GDT Res %) and the phylogenic distance (PHYLO_DIST), both harnessed from Table 8 and Table 1 respectively. We also evaluated Table 5 to screen the residue percentages altered by more than 0.2 against the Highest_TM and Lowest_TM structural matches for a particular protein and represented them as Letter Y (i.e. Yes). The Table 10 also enlists an average percentage alternation of a particular residue as "Percent Change". It further shows that the amino acids alanine, tyrosine and phenylalanine are the most frequently changed or evolutionarily altered amino acids (79.4%) and similarly the amino acids glycine, arginine, aspartate, leucine, serine, asparagine, valine, threonine, methionine and glutamine are also found to be significantly altered. We also observed that an evolutionary alteration percentage for histidine and glutamate residues is exactly 50% (Count of Y in the respective column) and such evolutionary variation fraction is even lower for proline, lysine, cysteine and tryptophan.
Here, we saw a particular trend that all such significant and comparatively lesser noteworthy evolutionary altered percentages of certain specific residues equally include several hydrophobic and hydrophilic amino acids. In this regard, for the fact that hydrophobic amino acids are usually present in the protein core to maintain the overall functional conformation, it is often presumed that such residues are rarely altered evolutionarily. However here we could examine that some of the hydrophobic residue percentages have changed quite significantly parallel to that of hydrophilic residues. We observed that 3NKJA-2RJYA structural comparison showed a GDT residue, TM_Score similarity of 100% and 0.95413 respectively, despite having a non-zero phylogenetic distance (PHYLO_DIST) of 0.0197. This can be fairly attributed to alteration of a few specific amino acids, as represented by leucine and glycine (Shown as Y in Table 10).
Similarly we observed that 1YU7X-1YU8X distance was lesser than 1YU8X-2RJYA distance which clearly implies that the former pair TM_Score is higher than the latter one, although in our analysis it gave quite contrary results. Comparing the count of altered amino acids for both these pairs, it was found higher for 1YU7X-1YU8X than 1YU8X- 2RJYA (4/17=23.529% and 2/17=11.767% respectively) and it thus implies that former pair should have a comparatively lower TM_Score, being 0.99097 and 0.99691 for the former and latter pairs respectively. This data was completely synchronous with our observation, as enlisted in Table 10. It further implies that the former pair with higher amino acid percentage alteration should show a higher phylogenetic distance while to our surprise; it was exactly the other way round. This indirectly means that phylogenetic distance as such is an incomplete term to compare the similarity of two proteins, as its sequence based analysis may prove to be wrong.
As per Table 10, we observe that 3NKJA-2RJYA phylogenetic distance is more than 1YU8X-2RJYA and exactly the same correlation was found through their TM_Score comparison, being 0.95413 and 0.99691 respectively for both these pairs. Further adding to it, 2RJXA-2RJWA and 3MYAA-2RJWA protein pairs showed a quite converse relationship. Here in these compared protein pairs, two amino acids tyrosine and histidine show significant evolutionary alteration for 2RJXA-2RJWA pair and such variant residues for 3MYAA-2RJWA pair are tyrosine and phenylalanine. As tyrosine is common among both these considered pairs, their TM_Score difference can be mainly and respectively attributed to the change in the availability percentage of histidine and phenylalanine residues and hence it further implies that the chemical nature of variant amino acid as well as its specific functional locus in a protein is extremely important.
Here in this entire analysis, we simply observe that the TM_Score is comparatively lower among the sequences extracted from different source organisms and it shows that the protein structures are relatively more conserved within the species, as earlier shown by Doolittle. In this phylogenetic distance data (Table 10) the GDT residue percentage and TM_Score figures are pretty high for the structures with zero phylogenetic distance, as expected. Comparing 2K6MS-2K6NS, we do not see any change in their encoded percentages of amino acids, although we still find that their GDT residue percentage is not equal to 100 while their TM_Score is also not equivalent to 1.
This observation could be reasonably attributed to interaction of such structures with different or same ligands in varying microenvironment available in same/different source organisms, or it might also be the result of minor sequence/structural shifts for certain specific residues, or the evolutionary extension/shortening of protein sequences despite retaining the earlier percentages of all the encoded amino acids. Furthermore we observe that the Lowest_TM Score match is still more than 0.5 even when their PHYLO_DIST distance is reasonably good and it highlights the structural conservation of protein structures. In Table 10, we logically observe that several residue percentages are significantly altered for the protein pairs with a low TM_Score. Quite intriguingly, the pairs including 3MYCA-2RJVA with variant residue percentages also show minimal phylogenetic distance and a considerable TM_Score.
We observe that functional protein copies of different organisms, with almost similar overall topology, show a widely ranged amino acid percentage variation. To avoid the complication in study, we have not considered the evolutionary alterations at the level of DNA which could have resulted in silent mutations without altering the encoded amino acid at that specific position. Thus evolutionary alterations only varying the encoded native amino acid are considered significant here.
Considering two proteins lying far apart in the phylogenetic tree, we normally assume them being significantly different, both in terms of sequence and structural similarity. And we have proven this scientific myth to be wrong here after showing several such instances where the phylogenetically distant proteins show TM_Score more than 0.5 and a significant structural similarity. Quite interestingly, the phylogenetic distance among the proteins belonging to the same species is found to be lesser than that of inter-species protein comparisons. Similarly, an amino acid frequency is found to be similar within the proteins belonging to the same species.
Regarding our predictions about the phylogenetic trees and their further respective analytical observation, we find that it is totally inefficient to extract the correct level of similarity among protein sequences. Simply relying on the sequence information based phylogenetic tree, we should not categorize proteins to be evolutionarily linked or divergent. Hence when the structural information exists or can be predicted for protein sequences, we should not restrict ourselves to the sequence based phylogenetic analysis. It is because phylogenetically distant proteins need not be structurally dissimilar also, as shown by our villin HP structural comparison study.
Intra-species structural comparison analysis for the villin HP proteins show that protein conformation is extensively conserved and the observed marginal structural shift is generally due to the altered amino acids or differential availability of certain specific ligand molecules in the immediate micro-environment. It is also well observed that sequences encoding the comprehensively altered amino acid percentages normally show a low structural similarity in terms of TM_Score. As per our detailed analysis of the residue percentage comparison across functionally similar proteins of different source organisms, tryptophan is found to be the least altering and it thus advocates the vital role of tryptophan to maintain the structural topology of the villin HP. It also shows that tryptophan is not extensively altered evolutionarily to preserve the major structural topology. Similarly alanine, tyrosine and phenylalanine are found to be the mostly variant amino acids across the selected villin HP proteins. We normally argue that hydrophobic amino acids are mostly buried in the protein core and they do not show extensive evolutionary variations. However as per our observations the percentage change of hydrophobic and hydrophilic amino acids stands almost similar. It is thus well realized through this study that the count and physicochemical nature of altered amino acids in a protein sequence proportionally imply the alteration in its conformation.
Hence we can say that, the amino acids with significantly altered percentages are the ones which are less important for the protein structure (including the original primary function) and are evolutionarily more preferably chosen than other amino acids. The evolutionarily unaltered amino acids should thus be the ones which are more conserved, being important for the functional stability and structure maintenance. So, it seems obvious that the amino acids which are evolutionarily altered in a protein are the ones which give an additional functional edge to the protein conformation to attain an increased functionality or the half-life in the exposed micro-environmental constraints. Hence we can state that the protein sequences normally alter to a great extent during evolution, but they can still retain their overall structural topology to maintain the native function.
Through the comprehensively analyzed data and contemplating the enlisted as well as implied information, we conclude that several residue substituting mutations can normally occur in protein sequences without altering their function and overall topology. Although this study was done on villin HP, it can be equally extrapolated to other proteins as well. Here it is well realized that the change in percentage of an amino acid can vary within a wide range across several similar functional copies available in same or different species, but the overall structure may still be unerringly similar retaining its native function. Furthermore, nature alters the functionally important amino acids to improve their specific role in the evolved conformation which is still almost similar to the native structure. Likewise nature alters structurally insignificant amino acids also to provide the altered functionality to the evolved conformation, possibly due to binding of some new ligands.
We also conclude that the phylogenetic tree single-handedly cannot extract the detailed similarity among the protein sequences. Hence, a better phylogenetic model predicting the structure of considered protein sequences is essentially needed to reliably find the significant evolutionary or functional relationship information among the considered protein sequences. Such structural comparison guided highly informative evolutionary tree would thus be far better than the routinely used sequence based phylogenetic trees.
Although the robustness of protein structures is well understood, we often assume that evolved sequences might have altered conformations. However precisely concluding, this study illustrates that the protein structures or their functional domains are not evolutionarily robust over every residue substitution. Or in other words, nature specifically tweaks certain amino acids in a protein domain for attaining its desired function in its constrained micro- environment. To predict this structural robustness simply from the protein sequence, we can specifically employ the characteristics of the altered residues along with the provided proximal sequence and structural context. Normally, we assume that a protein is evolutionarily susceptible for a few residues and they are responsible for the evolved functionalities of its conformation. However, all these susceptible residues do not evolve quite equally likely to any other different residue. We should thus predict the consequence of such an alteration so that the evolutionarily related information of every single protein residue can be correctly mapped to make us competent enough to reliably select even the distantly related templates for correctly modelling a protein sequence. Although this information is worked out in HMM profile based template search algorithms, they fail to extrapolate it to every single target residue and they do not efficiently employ the evolutionarily related, functionally significant information of a residue along with the other residues that are within a predefined residue boundary cutoff. It is because all such proximal residues within a defined structural boundary cutoff are responsible for the functionally positive evolutionary alteration of a single one. Hence, if we properly track the evolutionarily nature and structural implication of the altered residues, we can reliably link the distant relationship of a protein to its related orthologous that are conventionally not considered.
Further concluding, the application of this study is its exquisite importance for protein structure prediction methodologies which search homologous as well as reliable templates for modelling a protein sequence. As per this study, a phylogenetically distant sequence may still share a similar structure and we should try finding such structurally solved conformations available for modelling a target sequence. During the routinely employed template search methodology, by plausibly improving the substitution scores for the computed same-column template profile residues retaining a good reliable TM_Score in their structural comparison, along with a maximal reliable span of the considered target sequence with minimal count of such selected templates, we might reach closer to our ultimate goal to quickly as well as efficiently search evolutionarily distant and reliable hits for modelling an improved near- native conformation of a target sequence.

![]()

 |
| Figure 1: Flowchart representing the overview of the complete methodology used in the research. |
 |
| Figure 2: Phylogenetic tree for Headpiece. |
 |
| Figure 3: Phylogenetic tree for Sub-domain. |
 |
| Figure 4: Phylogenetic tree for Headpiece + Sub-domain. |
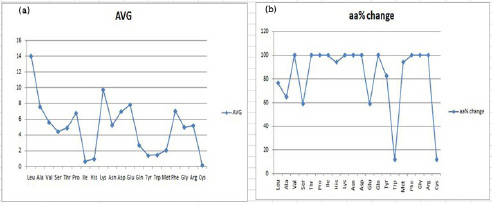 |
| Figure 5: (a) Graph for the average occurrence of amino acid residues in HP structures (b) Percentage variation in occurrence of amino acid residues in HP structures. |
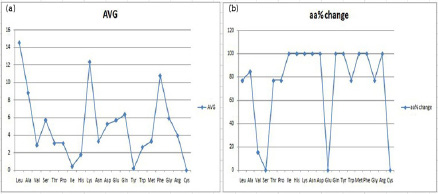 |
| Figure 6: (a) Graph for the average occurrence of amino acid residues in the SD structures (b) Percentage variation in occurrence of amino acid residues in SD structures. |
| PHYO_DIST HP | 1QZPA | 1ZV6A | 1UJSA | 2K6MS | 2K6NA | 2RJVA | 2RJWA | 3MYAA | 3MYCA | 1YU5X | 1YU8X | 2RJXA | 3NKJA | 1QQVA | 2RJYA | 3MYEX | 1YU7X |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1QZPA | 0 | 0.02057 | 0.0748 | 0.00332 | 0.00332 | 0.84188 | 0.84188 | 0.82264 | 0.82264 | 0.8438 | 0.82432 | 0.8438 | 0.8241 | 0.8438 | 0.8438 | 0.8438 | 0.8247 |
| 1ZV6A | 0 | 0.09537 | 0.01725 | 0.01725 | 0.86245 | 0.86245 | 0.84321 | 0.84321 | 0.86437 | 0.84489 | 0.86437 | 0.84467 | 0.86437 | 0.86437 | 0.86437 | 0.84527 | |
| 1UJSA | 0 | 0.078 12 | 0.07812 | 0.76708 | 0.76708 | 0.74784 | 0.74784 | 0.769 | 0.74952 | 0.769 | 0.7493 | 0.769 | 0.769 | 0.769 | 0.7499 | ||
| 2K6MS | 0 | 0 | 0.8452 | 0.8452 | 0.82596 | 0.82596 | 0.84712 | 0.82728 | 0.84712 | 0.82742 | 0.84712 | 0.84712 | 0.84712 | 0.82802 | |||
| 2K6NA | 0 | 0.8452 | 0.8452 | 0.82596 | 0.82596 | 0.84712 | 0.82728 | 0.84712 | 0.82742 | 0.84712 | 0.84712 | 0.84712 | 0.82802 | ||||
| 2RJVA | 0 | 0 | 0.01924 | 0.01924 | 0.00192 | 0.01756 | 0.00192 | 0.01778 | 0.00192 | 0.00192 | 0.00192 | 0.01718 | |||||
| 2RJWA | 0 | 0.01924 | 0.01924 | 0.00192 | 0.01756 | 0.00192 | 0.01778 | 0.00192 | 0.00192 | 0.00192 | 0.01718 | ||||||
| 3MYAA | 0 | 0 | 0.02116 | 0.02116 | 0.00146 | 0.02116 | 0.02116 | 0.02116 | 0.02116 | 0.00206 | |||||||
| 3MYCA | 0 | 0.02116 | 0.02116 | 0.00146 | 0.02116 | 0.02116 | 0.02116 | 0.02116 | 0.00206 | ||||||||
| 1YU5X | 0 | 0.01948 | 0 | 0.0197 | 0 | 0 | 0 | 0.0191 | |||||||||
| 1YU8X | 0 | 0.01948 | 0.00022 | 0.01948 | 0.01948 | 0.01948 | 0.00038 | ||||||||||
| 2RJXA | 0 | 0.0197 | 0 | 0 | 0 | 0.0191 | |||||||||||
| 3NKJA | 0 | 0.0197 | 0.0197 | 0.0197 | 0.0006 | ||||||||||||
| 1QQVA | 0 | 0 | 0 | 0.0191 | |||||||||||||
| 2RJYA | 0 | 0 | 0.0191 | ||||||||||||||
| 3MYEX | 0 | 0.0191 | |||||||||||||||
| 1YU7X | 0 | ||||||||||||||||
| Table 1: Phylogenetic distances for Headpiece | |||||||||||||||||
| Gallus gallus | Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1QQVA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 1.49254 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 1VIIA | 13.8889 | 8.33333 | 2.77778 | 5.55556 | 2.77778 | 2.77778 | 0 | 0 | 13.8889 | 5.55556 | 5.55556 | 5.55556 | 5.55556 | 0 | 2.77778 | 5.55556 | 11.1111 | 5.55556 | 2.77778 | 0 |
| 1YU5X | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 1.49254 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 1YU7X | 14.0625 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 1.5625 | 0 | 1.5625 | 7.8125 | 4.6875 | 4.6875 | 0 |
| 1YU8X | 14.0625 | 9.375 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 4.6875 | 3.125 | 0 |
| 2RJVA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 0 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 1.49254 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 2RJWA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 0 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 1.49254 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 2RJXA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 1.49254 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 2RJYA | 14.0625 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 4.6875 | 4.6875 | 0 |
| 3MYAA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 0 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 8.95522 | 4.47761 | 4.47761 | 0 |
| 3MYCA | 14.0625 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 0 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 9.375 | 4.6875 | 4.6875 | 0 |
| 3MYEX | 12.5 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 6.25 | 4.6875 | 0 |
| 3NKJA | 12.5 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 6.25 | 4.6875 | 0 |
| Homo sapiens | Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys |
| 1QZPA | 13.2353 | 5.88235 | 5.88235 | 5.88235 | 2.94118 | 7.35294 | 1.47059 | 1.47059 | 10.2941 | 2.94118 | 2.94118 | 8.82353 | 1.47059 | 1.49254 | 1.47059 | 1.49254 | 5.88235 | 5.88235 | 8.82353 | 0 |
| 1UJSA | 11.3636 | 3.40909 | 4.54545 | 10.2273 | 4.54545 | 3.40909 | 2.27273 | 1.13636 | 5.68182 | 4.54545 | 3.40909 | 7.95455 | 3.40909 | 4.54545 | 2.27273 | 2.27273 | 4.54545 | 9.09091 | 11.3636 | 0 |
| 1ZV6A | 13.2353 | 5.88235 | 5.88235 | 4.41176 | 2.94118 | 7.35294 | 1.47059 | 1.47059 | 10.2941 | 2.94118 | 2.94118 | 10.2941 | 1.47059 | 2.94118 | 1.47059 | 4.41176 | 5.88235 | 5.88235 | 8.82353 | 0 |
| 2K6MS | 19.403 | 10.4478 | 2.98507 | 0 | 4.47761 | 7.46269 | 2.98507 | 0 | 10.4478 | 2.98507 | 8.95522 | 7.46269 | 1.49254 | 4.47761 | 1.49254 | 2.98507 | 4.47761 | 2.98507 | 2.98507 | 1.49254 |
| 2K6NA | 19.403 | 10.4478 | 2.98507 | 0 | 4.47761 | 7.46269 | 2.98507 | 0 | 10.4478 | 2.98507 | 8.95522 | 7.46269 | 1.49254 | 4.47761 | 1.49254 | 2.98507 | 4.47761 | 2.98507 | 2.98507 | 1.49254 |
| Synthetic construct (Homo sapiens) | Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys |
| 1UNCA | 11.4286 | 11.4286 | 0 | 5.71429 | 5.71429 | 5.71429 | 2.85714 | 0 | 11.4286 | 2.85714 | 2.85714 | 5.71429 | 8.57143 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 1UNDA | 11.1111 | 5.55556 | 5.55556 | 5.55556 | 2.77778 | 2.77778 | 2.77778 | 0 | 11.1111 | 0 | 2.77778 | 5.55556 | 11.1111 | 2.77778 | 2.77778 | 2.77778 | 11.1111 | 11.1111 | 2.77778 | 0 |
| Synthetic construct | Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys |
| 1WY3A | 17.1429 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 11.4286 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.8514 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 1WY4A | 17.1429 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 11.4286 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 1YRFA | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 14.2857 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 1YRIA | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 14.2857 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 2JM0A | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 0 | 0 | 5.71429 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 17.1429 | 0 |
| 2PPZA | 16.6667 | 8.33333 | 2.77778 | 5.55556 | 2.77778 | 2.77778 | 0 | 0 | 13.8889 | 5.55556 | 5.55556 | 5.55556 | 5.55556 | 0 | 2.77778 | 5.55556 | 11.1111 | 2.77778 | 2.77778 | 0 |
| Synthetic construct (Gallus gallus) | Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys |
| 3TJWA | 14.7059 | 8.82353 | 2.94118 | 5.88235 | 2.94118 | 2.94118 | 0 | 2.94118 | 14.7059 | 2.94118 | 5.88235 | 5.88235 | 5.88235 | 0 | 2.94118 | 2.94118 | 8.82353 | 5.88235 | 2.94118 | 0 |
| 3TRVA | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 14.2857 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 3TRWA | 14.7059 | 11.7647 | 2.94118 | 5.88235 | 2.94118 | 2.94118 | 0 | 2.94118 | 14.7059 | 2.94118 | 5.88235 | 5.88235 | 5.88235 | 0 | 0 | 2.94118 | 8.82353 | 5.88235 | 2.94118 | 0 |
| 3TRYA | 14.7059 | 8.82353 | 2.94118 | 5.88235 | 2.94118 | 2.94118 | 0 | 2.94118 | 14.7059 | 2.94118 | 5.88235 | 5.88235 | 5.88235 | 0 | 2.94118 | 2.94118 | 8.82353 | 5.88235 | 2.94118 | 0 |
| Table 2: Amino acid percentage distribution according to the source of PDB filess | ||||||||||||||||||||
| PHYLO_DIST SD | 1UNDA | 1UNCA | 3TJWA | 3TRYA | 2JM0A | 1VIIA | 2PPZA | 1YRFA | 3TRVA | 3TRWA | 1YRIA | 1WY3A | 1WY4A |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1UNDA | 0 | 0.1379 | 0.23546 | 0.23546 | 0.00005 | 0.18955 | 0.14125 | 0.23546 | 0.23546 | 0.18631 | 0.23546 | 0.18637 | 0.18637 |
| 1UNCA | 0 | 0.09756 | 0.09756 | 0.13795 | 0.05165 | 0.00335 | 0.09756 | 0.09756 | 0.04841 | 0.09756 | 0.04847 | 0.04847 | |
| 3TJWA | 0 | 0 | 0.23551 | 0.04591 | 0.09421 | 0 | 0 | 0.04915 | 0 | 0.04909 | 0.04909 | ||
| 3TRYA | 0 | 0.23551 | 0.04591 | 0.09421 | 0 | 0 | 0.04915 | 0 | 0.04909 | 0.04909 | |||
| 2JM0A | 0 | 0.1896 | 0.1413 | 0.23551 | 0.23551 | 0.18636 | 0.23551 | 0.18642 | 0.18642 | ||||
| 1VIIA | 0 | 0.0483 | 0.04591 | 0.04591 | 0.00324 | 0.04591 | 0.00318 | 0.00318 | |||||
| 2PPZA | 0 | 0.09421 | 0.09421 | 0.04506 | 0.09421 | 0.04512 | 0.00318 | ||||||
| 1YRFA | 0 | 0 | 0.04915 | 0 | 0.04909 | 0.04909 | |||||||
| 3TRVA | 0 | 0.04915 | 0 | 0.04909 | 0.04909 | ||||||||
| 3TRWA | 0 | 0.04915 | 0.00006 | 0.00006 | |||||||||
| 1YRIA | 0 | 0.04909 | 0.04909 | ||||||||||
| 1WY3A | 0 | 0 | |||||||||||
| 1WY4A | 0 | ||||||||||||
| Table 3: Phylogenetic distances for Sub-domain | |||||||||||||
| PHYLO_DISTHP+SD | 1QZPA | 1ZV6A | 1UJSA | 2PPZA | 3MYCA | 3MYEX | 2JM0A | 3MYAA | 2RJWA | 2RJXA | 2RJVA | 3NKJA | 1QQVA | 2K6MS | 2K6NA | 1UNDA | 1UNCA | 1YU8X | 1YU5X | 1VIIA | 3TRVA | 3TRYA | 3TRWA | 3TJWA | 1WY3A | 1WY4A | 1YRFA | 1YRIA | 1YU7X | 2RJYA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1QZPA | 0 | 0.07338 | 0.03001 | 0.30366 | 0.38186 | 0.38186 | 0.07846 | 0.38186 | 0.38186 | 0.38186 | 0.38186 | 0.30248 | 0.38186 | 0.00539 | 0.00539 | 0.07329 | 0.30991 | 0.38186 | 0.38186 | 0.38186 | 0.30964 | 0.30964 | 0.22944 | 0.30964 | 0.22947 | 0.22947 | 0.30964 | 0.30964 | 0.31178 | 0.38186 |
| 1ZV6A | 0 | 0.04337 | 0.37704 | 0.45524 | 0.45524 | 0.15184 | 0.45524 | 0.45524 | 0.45524 | 0.45524 | 0.37586 | 0.45524 | 0.06799 | 0.06799 | 0.00009 | 0.38329 | 0.45524 | 0.45524 | 0.45524 | 0.38302 | 0.38302 | 0.30282 | 0.38302 | 0.30285 | 0.30285 | 0.38302 | 0.38302 | 0.38516 | 0.45524 | |
| 1UJSA | 0 | 0.33367 | 0.41187 | 0.41187 | 0.10847 | 0.41187 | 0.41187 | 0.41187 | 0.41187 | 0.33249 | 0.41187 | 0.02462 | 0.02462 | 0.04328 | 0.33992 | 0.41187 | 0.41187 | 0.41187 | 0.33965 | 0.33965 | 0.25945 | 0.33965 | 0.25948 | 0.25948 | 0.33965 | 0.33965 | 0.34179 | 0.41187 | ||
| 2PPZA | 0 | 0.0782 | 0.0782 | 0.2252 | 0.0782 | 0.0782 | 0.0782 | 0.0782 | 0.00118 | 0.0782 | 0.30905 | 0.30905 | 0.37695 | 0.00625 | 0.0782 | 0.0782 | 0.0782 | 0.00598 | 0.00598 | 0.07422 | 0.00598 | 0.07419 | 0.07419 | 0.00598 | 0.00598 | 0.00812 | 0.0782 | |||
| 3MYCA | 0 | 0 | 0.3034 | 0 | 0 | 0 | 0 | 0 | 0 | 0.38725 | 0.38725 | 0.45515 | 0.07195 | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | ||||
| 3MYEX | 0 | 0.3034 | 0 | 0 | 0 | 0 | 0 | 0 | 0.38725 | 0.38725 | 0.45515 | 0.07195 | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | |||||
| 2JM0A | 0.3034 | 0.3034 | 0.3034 | 0.3034 | 0.22402 | 0.3034 | 0.08385 | 0.08385 | 0.15175 | 0.23145 | 0.3034 | 0.3034 | 0.3034 | 0.23118 | 0.23118 | 0.15098 | 0.23118 | 0.15101 | 0.15101 | 0.23118 | 0.23118 | 0.23332 | 0.3034 | |||||||
| 3MYAA | 0 | 0 | 0 | 0 | 0.07938 | 0 | 0.38725 | 0.38725 | 0.45515 | 0.07195 | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | |||||||
| 2RJWA | 0 | 0 | 0 | 0.07938 | 0 | 0.38725 | 0.38725 | 0.45515 | 0.07195 | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | ||||||||
| 2RJXA | 0 | 0 | 0.07938 | 0 | 0.38725 | 0.38725 | 0.45515 | 0.07195 | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | |||||||||
| 2RJVA | 0 | 0.07938 | 0 | 0.38725 | 0.38725 | 0.45515 | 0.07195 | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | ||||||||||
| 3NKJA | 0 | 0.07938 | 0.08385 | 0.08385 | 0.37577 | 0.00743 | 0.07938 | 0.07938 | 0.07938 | 0.00716 | 0.00716 | 0.07304 | 0.00716 | 0.07301 | 0.07301 | 0.23118 | 0.23118 | 0.0093 | 0.07938 | |||||||||||
| 1QQVA | 0 | 0.38725 | 0.38725 | 0.45515 | 0.07195 | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | ||||||||||||
| 2K6MS | 0 | 0 | 0.0679 | 0.3153 | 0.38725 | 0.38725 | 0.38725 | 0.31503 | 0.31503 | 0.23483 | 0.31503 | 0.23486 | 0.23486 | 0.31503 | 0.31503 | 0.31717 | 0.38725 | |||||||||||||
| 2K6NA | 0 | 0.0679 | 0.3153 | 0.38725 | 0.38725 | 0.38725 | 0.31503 | 0.31503 | 0.23483 | 0.31503 | 0.23486 | 0.23486 | 0.31503 | 0.31503 | 0.31717 | 0.38725 | ||||||||||||||
| 1UNDA | 0 | 0.3832 | 0.45515 | 0.45515 | 0.45515 | 0.38293 | 0.38293 | 0.30273 | 0.38293 | 0.30276 | 0.30276 | 0.38293 | 0.38293 | 0.38507 | 0.45515 | |||||||||||||||
| 1YU8X | 0 | 0.07195 | 0.07195 | 0.07195 | 0.00027 | 0.00027 | 0.08047 | 0.00027 | 0.08044 | 0.08044 | 0.00027 | 0.00027 | 0.00187 | 0.07195 | ||||||||||||||||
| 1YU5X | 0 | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | |||||||||||||||||
| 1VIIA | 0 | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | ||||||||||||||||||
| 3TRVA | 0 | 0.07222 | 0.07222 | 0.15242 | 0.07222 | 0.15239 | 0.15239 | 0.07222 | 0.07222 | 0.07008 | 0 | |||||||||||||||||||
| 3TRYA | 0 | 0 | 0.0802 | 0 | 0.08017 | 0.08017 | 0 | 0 | 0.00214 | 0.07222 | ||||||||||||||||||||
| 3TRWA | 0 | 0.0802 | 0 | 0.08017 | 0.08017 | 0 | 0 | 0.00214 | 0.07222 | |||||||||||||||||||||
| 3TJWA | 0 | 0.0802 | 0.00003 | 0.00003 | 0.0802 | 0.0802 | 0.08234 | 0.15242 | ||||||||||||||||||||||
| 1WY3A | 0 | 0.08017 | 0.08017 | 0 | 0 | 0.00214 | 0.07222 | |||||||||||||||||||||||
| 1WY4A | 0 | 0 | 0.08017 | 0.08017 | 0.08231 | 0.15239 | ||||||||||||||||||||||||
| 1YRFA | 0 | 0.08017 | 0.08017 | 0.08231 | 0.15239 | |||||||||||||||||||||||||
| 1YRIA | 0 | 0 | 0.00214 | 0.07222 | ||||||||||||||||||||||||||
| 1YU7X | 0 | 0.00214 | 0.07222 | |||||||||||||||||||||||||||
| 2RJYA | 0 | 0.07008 | ||||||||||||||||||||||||||||
| Table 4: Phylogenetic distances for Headpiece + Sub-domain | 0 | |||||||||||||||||||||||||||||
| HP amino acid% | Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys |
| 1QQVA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 1.49254 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1QZPA | 13.2353 | 5.88235 | 5.88235 | 5.88235 | 2.94118 | 7.35294 | 1.47059 | 1.47059 | 10.2941 | 2.94118 | 2.94118 | 8.82353 | 1.47059 | 2.94118 | 1.47059 | 4.41176 | 5.88235 | 5.88235 | 8.82353 | 0 |
| 1UJSA | 11.3636 | 3.40909 | 4.54545 | 10.2273 | 4.54545 | 3.40909 | 2.27273 | 1.13636 | 5.68182 | 4.54545 | 3.40909 | 7.95455 | 3.40909 | 4.54545 | 2.27273 | 2.27273 | 4.54545 | 9.09091 | 11.3636 | 0 |
| 1YU7X | 14.0625 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 1.5625 | 0 | 1.5625 | 7.8125 | 4.6875 | 4.6875 | 0 |
| 1YU8X | 14.0625 | 9.375 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 4.6875 | 3.125 | 0 |
| 1YU5X | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 1.49254 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 1ZV6A | 13.2353 | 5.88235 | 5.88235 | 4.41176 | 2.94118 | 7.35294 | 1.47059 | 1.47059 | 10.2941 | 2.94118 | 2.94118 | 10.2941 | 1.47059 | 2.94118 | 1.47059 | 4.41176 | 5.88235 | 5.88235 | 8.82353 | 0 |
| 2K6MS | 19.403 | 10.4478 | 2.98507 | 0 | 4.47761 | 7.46269 | 2.98507 | 0 | 10.4478 | 2.98507 | 8.95522 | 7.46269 | 1.49254 | 4.47761 | 1.49254 | 2.98507 | 4.47761 | 2.98507 | 2.98507 | 1.49254 |
| 2K6NA | 19.403 | 10.4478 | 2.98507 | 0 | 4.47761 | 7.46269 | 2.98507 | 0 | 10.4478 | 2.98507 | 8.95522 | 7.46269 | 1.49254 | 4.47761 | 1.49254 | 2.98507 | 4.47761 | 2.98507 | 2.98507 | 1.49254 |
| 2RJVA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 0 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 1.49254 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 2RJWA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 0 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 1.49254 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 2RJXA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 1.49254 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 7.46269 | 4.47761 | 4.47761 | 0 |
| 2RJYA | 14.0625 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 4.6875 | 4.6875 | 0 |
| 3MYAA | 13.4328 | 7.46269 | 5.97015 | 4.47761 | 5.97015 | 7.46269 | 0 | 0 | 10.4478 | 5.97015 | 7.46269 | 7.46269 | 2.98507 | 0 | 1.49254 | 1.49254 | 8.95522 | 4.47761 | 4.47761 | 0 |
| 3MYCA | 14.0625 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 0 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 9.375 | 4.6875 | 4.6875 | 0 |
| 3MYEX | 12.5 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 6.25 | 4.6875 | 0 |
| 3NKJA | 12.5 | 7.8125 | 6.25 | 4.6875 | 4.6875 | 6.25 | 0 | 1.5625 | 9.375 | 6.25 | 7.8125 | 7.8125 | 3.125 | 0 | 1.5625 | 1.5625 | 7.8125 | 6.25 | 4.6875 | 0 |
| AVG | 14.0287 | 7.60488 | 5.6236 | 4.44189 | 4.9017 | 6.78332 | 0.65789 | 0.9628 | 9.77071 | 5.27758 | 6.99135 | 7.86169 | 2.70563 | 1.40768 | 1.46863 | 2.08215 | 7.05715 | 4.99656 | 5.20053 | 0.17559 |
| aa% change | 76.4706 | 64.7059 | 100 | 58.8235 | 100 | 100 | 100 | 94.1176 | 100 | 100 | 100 | 58.8235 | 100 | 82.3529 | 11.7647 | 94.1176 | 100 | 100 | 100 | 11.7647 |
Table 5: Percentage of amino acid residues for Headpiece |
||||||||||||||||||||
| SD amino acid% | Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys |
| 1UNCA | 11.4286 | 11.4286 | 0 | 5.71429 | 5.71429 | 5.71429 | 2.85714 | 0 | 11.4286 | 2.85714 | 2.85714 | 5.71429 | 8.57143 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1UNDA | 11.1111 | 5.55556 | 5.55556 | 5.55556 | 2.77778 | 2.77778 | 2.77778 | 0 | 11.1111 | 0 | 2.77778 | 5.55556 | 11.1111 | 2.77778 | 2.77778 | 2.77778 | 11.1111 | 11.1111 | 2.77778 | 0 |
| 1VIIA | 13.8889 | 8.33333 | 2.77778 | 5.55556 | 2.77778 | 2.77778 | 0 | 0 | 13.8889 | 5.55556 | 5.55556 | 5.55556 | 5.55556 | 0 | 2.77778 | 5.55556 | 11.1111 | 5.55556 | 2.77778 | 0 |
| 1WY3A | 17.1429 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 11.4286 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 1WY4A | 17.1429 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 11.4286 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 1YRFA | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 14.2857 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 1YRIA | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 14.2857 | 2.85714 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 2JM0A | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 0 | 0 | 5.71429 | 5.71429 | 5.71429 | 5.71429 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 17.1429 | 0 |
| 2PPZA | 16.6667 | 8.33333 | 2.77778 | 5.55556 | 2.77778 | 2.77778 | 0 | 0 | 13.8889 | 5.55556 | 5.55556 | 5.55556 | 5.55556 | 0 | 2.77778 | 5.55556 | 11.1111 | 2.77778 | 2.77778 | 0 |
| 3TJWA | 14.7059 | 8.82353 | 2.94118 | 5.88235 | 2.94118 | 2.94118 | 0 | 2.94118 | 14.7059 | 2.94118 | 5.88235 | 5.88235 | 5.88235 | 0 | 2.94118 | 2.94118 | 8.82353 | 5.88235 | 2.94118 | 0 |
| 3TRVA | 14.2857 | 8.57143 | 2.85714 | 5.71429 | 2.85714 | 2.85714 | 0 | 2.85714 | 14.2857 | 2.85714 | 5.71429 | 5.71429 | 5.7129 | 0 | 2.85714 | 2.85714 | 11.4286 | 5.71429 | 2.85714 | 0 |
| 3TRWA | 14.7059 | 11.7647 | 2.94118 | 5.88235 | 2.94118 | 2.94118 | 0 | 2.94118 | 14.7059 | 2.94118 | 5.88235 | 5.88235 | 5.88235 | 0 | 0 | 2.94118 | 8.82353 | 5.88235 | 2.94118 | 0 |
| 3TRYA | 14.7059 | 8.82353 | 2.94118 | 5.88235 | 2.94118 | 2.94118 | 0 | 2.94118 | 14.7059 | 2.94118 | 5.88235 | 5.88235 | 5.88235 | 0 | 2.94118 | 2.94118 | 8.82353 | 5.88235 | 2.94118 | 0 |
| AVG | 14.5109 | 8.80701 | 2.85212 | 5.71644 | 3.078 | 3.078 | 0.43346 | 1.77763 | 12.3192 | 3.29168 | 5.28298 | 5.71644 | 6.36357 | 0.21368 | 2.63198 | 3.28557 | 10.7541 | 5.93012 | 3.95712 | 0 |
| aa% change | 76.9231 | 84.6154 | 15.3846 | 0 | 76.9231 | 76.9231 | 100 | 100 | 100 | 100 | 100 | 0 | 100 | 100 | 76.9231 | 100 | 100 | 76.9231 | 100 | 0 |
| Table 6: Percentage of amino acid residues for Sub-domain | ||||||||||||||||||||
| Our Work | (Cornish-Bowden, A., 1983 | (Doolittle, R.F., 1989 ) | (Carugo O., 2008) | ||||
| AVG-HP | AVG-SD | AVG-118 | AVG-1150 | AVG_0.55_million | Substantially Variant for HP | Substantially Variant for SD | |
| Leu | 14.02865939 | 14.5108815 | 7.92 | 9.1 | 9.7 | 4.3286 | 4.8108 |
| Ala | 7.60487874 | 8.807009983 | 8.45 | 7.8 | 8 | 0.3951 | 0.807 |
| Val | 5.623600306 | 2.852115205 | 6.81 | 6.6 | 6.25 | 0.6264 | 3.3978 |
| Ser | 4.441886001 | 5.716440422 | 6.65 | 6.8 | 7.125 | 2.6831 | 1.4086 |
| Thr | 4.901701582 | 3.078000431 | 5.72 | 5.9 | 5.25 | 0.3483 | 2.172 |
| Pro | 6.783321518 | 3.078000431 | 4.53 | 5.2 | 4.8 | 1.9833 | 1.722 |
| Ile | 0.65788547 | 0.433455433 | 4.63 | 5.3 | 6.075 | 5.4171 | 5.6415 |
| His | 0.962803062 | 1.777634131 | 2.17 | 2.3 | 2.35 | 1.3872 | 0.57237 |
| Lys | 9.770714296 | 12.31918408 | 6.93 | 5.9 | 5.7 | 4.0707 | 6.61918 |
| Asn | 5.277579545 | 3.291675645 | 9.72 | 4.3 | 3.975 | 1.3025 | 1.9891 |
| Asp | 6.991353589 | 5.282984989 | 9.72 | 5.3 | 4.55 | 2.4413 | 0.73298 |
| Glu | 7.861687356 | 5.716440422 | 10.62 | 6.3 | 5.775 | 2.0866 | 0.05856 |
| Gln | 2.705634692 | 6.363571069 | 10.62 | 4.2 | 3.8 | 1.0943 | 2.56357 |
| Tyr | 1.407682706 | 0.213675214 | 3.32 | 3.2 | 2.9 | 1.4923 | 2.68633 |
| Trp | 1.468629544 | 2.631975867 | 1.31 | 1.4 | 1.4 | 0.0686 | 1.23197 |
| Met | 2.082154695 | 3.285570639 | 1.84 | 2.3 | 3.1 | 1.0178 | 0.18557 |
| Phe | 7.057149472 | 10.75414781 | 3.69 | 3.9 | 4.45 | 2.6071 | 6.30414 |
| Gly | 4.996555051 | 5.930115636 | 7.35 | 7.2 | 6.3 | 1.3034 | 0.36988 |
| Arg | 5.200530358 | 3.95712131 | 5.86 | 5.1 | 6.525 | 1.3244 | 2.63272 |
| Cys | 0.175592625 | 0 | 2.45 | 1.9 | 1.9 | 1.7244 | 1.9 |
Table 7: Comparison of the amino acid percentage present in our selected proteins with general amino acid percentage found in diverse sets of naturally occurring proteins |
|||||||
| TM_Score | 3NKJA | 1QQVA | 1QZPA | 1UJSA | 1YU5X | 1YU7X | 1YU8X | 1ZV6A | 2K6MS | 2K6NA | 2RJVA | 2RJWA | 2RJXA | 2RJYA | 3MYAA | 3MYCA | 3MYEX |
| 3NKJA | 1.00000 | ||||||||||||||||
| 1QQVA | 0.72342 | 1.00000 | |||||||||||||||
| 1QZPA | 0.70903 | 0.69753 | 1.00000 | ||||||||||||||
| 1UJSA | 0.60407 | 0.62027 | 0.61148 | 1.00000 | |||||||||||||
| 1YU5X | 0.91444 | 0.73989 | 0.73155 | 0.77349 | 1.00000 | ||||||||||||
| 1YU7X | 0.95383 | 0.74241 | 0.74729 | 0.78979 | 0.98861 | 1.00000 | |||||||||||
| 1YU8X | 0.95058 | 0.74004 | 0.74266 | 0.78461 | 0.99449 | 0.99097 | 1.00000 | ||||||||||
| 1ZV6A | 0.73974 | 0.79261 | 0.73323 | 0.79771 | 0.75820 | 0.74011 | 0.73370 | 1.00000 | |||||||||
| 2K6MS | 0.65319 | 0.65252 | 0.69062 | 0.69154 | 0.67720 | 0.66924 | 0.66108 | 0.69900 | 1.00000 | ||||||||
| 2K6NA | 0.63758 | 0.66049 | 0.68320 | 0.68642 | 0.67213 | 0.65615 | 0.64860 | 0.69357 | 0.90301 | 1.00000 | |||||||
| 2RJVA | 0.89670 | 0.71506 | 0.71879 | 0.75247 | 0.95697 | 0.94164 | 0.94179 | 0.74342 | 0.66172 | 0.65695 | 1.00000 | ||||||
| 2RJWA | 0.87204 | 0.74080 | 0.74917 | 0.76849 | 0.91103 | 0.89541 | 0.88948 | 0.78735 | 0.67405 | 0.66794 | 0.91323 | 1.00000 | |||||
| 2RJXA | 0.87235 | 0.73942 | 0.74493 | 0.76697 | 0.89604 | 0.89449 | 0.88695 | 0.77352 | 0.66843 | 0.66111 | 0.88700 | 0.95017 | 1.00000 | ||||
| 2RJYA | 0.95413 | 0.74396 | 0.74642 | 0.78923 | 0.99850 | 0.99003 | 0.99691 | 0.77490 | 0.68620 | 0.67275 | 0.98212 | 0.93080 | 0.92927 | 1.00000 | |||
| 3MYAA | 0.87088 | 0.75334 | 0.74578 | 0.77379 | 0.91173 | 0.89713 | 0.89006 | 0.79337 | 0.67863 | 0.67701 | 0.91075 | 0.99218 | 0.94954 | 0.89345 | 1.00000 | ||
| 3MYCA | 0.94219 | 0.74274 | 0.74305 | 0.77914 | 0.98244 | 0.98708 | 0.98868 | 0.76725 | 0.67788 | 0.66547 | 0.99534 | 0.93792 | 0.93298 | 0.98644 | 0.93878 | 1.00000 | |
| 3MYEX | 0.93842 | 0.74434 | 0.71620 | 0.76722 | 0.90525 | 0.90625 | 0.90523 | 0.74179 | 0.65597 | 0.63322 | 0.89305 | 0.88430 | 0.88781 | 0.90649 | 0.87957 | 0.89972 | 1.00000 |
| GDT Res% | 3NKJA | 1QQVA | 1QZPA | 1UJSA | 1YU5X | 1YU7X | 1YU8X | 1ZV6A | 2K6MS | 2K6NA | 2RJVA | 2RJWA | 2RJXA | 2RJYA | 3MYAA | 3MYCA | 3MYEX |
| 3NKJA | 100.00000 | ||||||||||||||||
| 1QQVA | 97.70992 | 100.00000 | |||||||||||||||
| 1QZPA | 96.96970 | 97.77778 | 100.00000 | ||||||||||||||
| 1UJSA | 82.89474 | 83.87097 | 87.17949 | 100.00000 | |||||||||||||
| 1YU5X | 97.70992 | 100.00000 | 97.77778 | 81.29032 | 100.00000 | ||||||||||||
| 1YU7X | 100.00000 | 97.70992 | 96.96970 | 80.26316 | 97.70992 | 100.00000 | |||||||||||
| 1YU8X | 100.00000 | 97.70992 | 96.96970 | 80.26316 | 97.70992 | 100.00000 | 100.00000 | ||||||||||
| 1ZV6A | 96.96970 | 96.29630 | 97.05882 | 85.89744 | 96.29630 | 95.45455 | 95.45455 | 100.00000 | |||||||||
| 2K6MS | 93.12977 | 91.04478 | 93.33333 | 81.29032 | 94.02985 | 93.12977 | 93.12977 | 93.33333 | 100.00000 | ||||||||
| 2K6NA | 93.12977 | 95.52239 | 93.33333 | 81.29032 | 95.52239 | 93.12977 | 93.12977 | 93.33333 | 98.50746 | 100.00000 | |||||||
| 2RJVA | 97.70992 | 98.50746 | 97.77778 | 81.29032 | 100.00000 | 97.70992 | 97.70992 | 96.29630 | 95.52239 | 95.52239 | 100.00000 | ||||||
| 2RJWA | 97.70992 | 97.01493 | 97.77778 | 81.29032 | 98.50746 | 97.70992 | 97.70992 | 96.29630 | 94.02985 | 95.52239 | 100.00000 | 100.00000 | |||||
| 2RJXA | 97.70992 | 95.52239 | 97.77778 | 78.70968 | 97.01493 | 97.70992 | 97.70992 | 94.81481 | 92.53731 | 91.04478 | 97.01493 | 97.01493 | 100.00000 | ||||
| 2RJYA | 100.00000 | 97.70992 | 96.96970 | 80.26316 | 97.70992 | 100.00000 | 100.00000 | 95.45455 | 93.12977 | 93.12977 | 97.70992 | 97.70992 | 97.70992 | 100.00000 | |||
| 3MYAA | 97.70992 | 97.01493 | 97.77778 | 81.29032 | 98.50746 | 97.70992 | 97.70992 | 96.29630 | 94.02985 | 95.52239 | 100.00000 | 100.00000 | 98.50746 | 97.70992 | 100.00000 | ||
| 3MYCA | 100.00000 | 97.70992 | 96.96970 | 80.26316 | 97.70992 | 100.00000 | 100.00000 | 95.45455 | 93.12977 | 93.12977 | 97.70992 | 97.70992 | 97.70992 | 100.00000 | 97.70992 | 100.00000 | |
| 3MYEX | 98.43750 | 96.18321 | 93.93939 | 80.26316 | 96.18321 | 96.87500 | 96.87500 | 93.93939 | 91.60305 | 91.60305 | 94.65649 | 96.18321 | 96.18321 | 96.87500 | 96.18321 | 96.87500 | 100.00000 |
Table 8: TM_Score and GDT residue percentage for Headpiece |
|||||||||||||||||
| TM_Score | 1UNCA | 1UNDA | 1VIIA | 1WY3A | 1WY4A | 1YRFA | 1YRIA | 2JM0A | 2PPZA | 3TJWA | 3TRVA | 3TRWA | 3TRYA |
| 1UNCA | 1.00000 | ||||||||||||
| 1UNDA | 0.58782 | 1.00000 | |||||||||||
| 1VIIA | 0.48480 | 0.53736 | 1.00000 | ||||||||||
| 1WY3A | 0.52442 | 0.72189 | 0.65591 | 1.00000 | |||||||||
| 1WY4A | 0.53226 | 0.69707 | 0.64216 | 0.90303 | 1.00000 | ||||||||
| 1YRFA | 0.57179 | 0.70510 | 0.67924 | 0.89612 | 0.91258 | 1.00000 | |||||||
| 1YRIA | 0.57148 | 0.64948 | 0.59500 | 0.72498 | 0.76147 | 0.79778 | 1.00000 | ||||||
| 2JM0A | 0.55877 | 0.56535 | 0.58616 | 0.67943 | 0.69899 | 0.81102 | 0.84125 | 1.00000 | |||||
| 2PPZA | 0.48763 | 0.49409 | 0.38994 | 0.55315 | 0.53775 | 0.50208 | 0.46147 | 0.41724 | 1.00000 | ||||
| 3TJWA | 0.31067 | 0.27579 | 0.27043 | 0.33189 | 0.34759 | 0.31710 | 0.32232 | 0.30996 | 0.30863 | 1.00000 | |||
| 3TRVA | 0.49679 | 0.62912 | 0.54321 | 0.77047 | 0.77403 | 0.72624 | 0.70945 | 0.58386 | 0.54648 | 0.30502 | 1.00000 | ||
| 3TRWA | 0.54534 | 0.72939 | 0.66434 | 0.95786 | 0.95476 | 0.94184 | 0.85670 | 0.70493 | 0.54875 | 0.33710 | 0.81582 | 1.00000 | |
| 3TRYA | 0.30721 | 0.28961 | 0.30513 | 0.32214 | 0.35113 | 0.29067 | 0.30528 | 0.28915 | 0.29086 | 0.95557 | 0.29846 | 0.34794 | 1.00000 |
| GDT Res% | 1UNCA | 1UNDA | 1VIIA | 1WY3A | 1WY4A | 1YRFA | 1YRIA | 2JM0A | 2PPZA | 3TJWA | 3TRVA | 3TRWA | 3TRYA |
| 1UNCA | 100.00000 | ||||||||||||
| 1UNDA | 95.7747 | 100.00000 | |||||||||||
| 1VIIA | 91.6667 | 88.8889 | 100.0000 | ||||||||||
| 1WY3A | 94.2857 | 90.1409 | 98.5916 | 100.0000 | |||||||||
| 1WY4A | 94.2857 | 90.1409 | 98.5916 | 100.0000 | 100.0000 | ||||||||
| 1YRFA | 97.1429 | 90.1409 | 98.5916 | 100.0000 | 100.0000 | 100.0000 | |||||||
| 1YRIA | 97.1429 | 92.9578 | 98.5916 | 100.0000 | 100.0000 | 100.0000 | 100.0000 | ||||||
| 2JM0A | 97.1429 | 92.9578 | 98.5916 | 100.0000 | 97.1429 | 100.0000 | 100.0000 | 100.0000 | |||||
| 2PPZA | 92.9578 | 91.6667 | 86.1111 | 84.5070 | 92.9578 | 95.7747 | 92.9578 | 92.9578 | 100.0000 | ||||
| 3TJWA | 72.4638 | 80.0000 | 68.5714 | 78.2609 | 81.1594 | 78.2609 | 78.2609 | 75.3623 | 71.4286 | 100.0000 | |||
| 3TRVA | 94.2857 | 90.1409 | 92.9578 | 100.0000 | 100.0000 | 100.0000 | 100.0000 | 100.0000 | 87.3239 | 84.0580 | 100.0000 | ||
| 3TRWA | 98.5507 | 91.4286 | 97.1429 | 98.5507 | 98.5507 | 98.5507 | 98.5507 | 98.5507 | 91.4286 | 85.2941 | 98.5507 | 100.0000 | |
| 3TRYA | 72.4638 | 71.4286 | 71.4286 | 78.2609 | 84.0580 | 75.3623 | 75.3623 | 72.4638 | 71.4286 | 97.0588 | 81.1594 | 82.3529 | 100.0000 |
Table 9: TM_Score and GDT residue percentage for Sub-domain |
|||||||||||||
| TM_Score | GDT Res% | PHYLO_DIST | EVOLUTIONARILY ALTERED AMINO ACIDS | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys | |||||
| 3NKJA | 3NKJA | 3NKJA | ||||||||||||||||||||||
| Highest_TM | 2RJYA | 0.95413 | 100 | 0.0197 | Y | Y | ||||||||||||||||||
| Lowest_TM | 1UJSA | 0.60407 | 82.89474 | 0.7493 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||
| 1QQVA | 1QQVA | 1QQVA | ||||||||||||||||||||||
| Highest_TM | 1ZV6A | 0.79261 | 96.29630 | 0.86437 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||||||
| Lowest_TM | 1UJSA | 0.62027 | 83.87097 | 0.769 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| 1QZPA | 1QZPA | 1QZPA | ||||||||||||||||||||||
| Highest_TM | 2RJWA | 0.74917 | 97.77778 | 0.84188 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||||
| Lowest_TM | 1UJSA | 0.61148 | 87.17949 | 0.0748 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| 1UJSA | 1UJSA | 1UJSA | ||||||||||||||||||||||
| Highest_TM | 1ZV6A | 0.79771 | 85.89744 | 0.09537 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Lowest_TM | 3NKJA | 0.60407 | 85.89744 | 0.7493 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||
| 1YU5X | 1YU5X | 1YU5X | ||||||||||||||||||||||
| Highest_TM | 2RJYA | 0.9985 | 97.70992 | 0 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||||
| Lowest_TM | 2K6NA | 0.67213 | 95.52239 | 0.84712 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||
| 1YU7X | 1YU7X | 1YU7X | ||||||||||||||||||||||
| Highest_TM | 1YU8X | 0.99097 | 100 | 0.00038 | Y | Y | Y | Y | ||||||||||||||||
| Lowest_TM | 2K6NA | 0.65615 | 93.12977 | 93.12977 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| 1YU8X | 1YU8X | 1YU8X | ||||||||||||||||||||||
| Highest_TM | 2RJYA | 0.99691 | 100 | 0.01948 | Y | Y | ||||||||||||||||||
| Lowest_TM | 2K6NA | 0.6486 | 93.12977 | 93.12977 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||
| 1ZV6A | 1ZV6A | 1ZV6A | ||||||||||||||||||||||
| Highest_TM | 1UJSA | 0.79771 | 85.89744 | 0.09537 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Lowest_TM | 2K6NA | 0.69357 | 93.33333 | 0.01725 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||||
| 2K6MS | 2K6MS | 2K6MS | ||||||||||||||||||||||
| Highest_TM | 2K6NA | 0.90301 | 98.50746 | 0 | ||||||||||||||||||||
| Lowest_TM | 1QQVA | 0.65252 | 91.04478 | 0.84712 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||
| 2K6NA | 2K6NA | 2K6NA | ||||||||||||||||||||||
| Highest_TM | 2K6MS | 0.90301 | 98.50746 | 0 | ||||||||||||||||||||
| Lowest_TM | 3MYEX | 0.63322 | 91.60305 | 0.84712 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| 2RJVA | 2RJVA | 2RJVA | ||||||||||||||||||||||
| Highest_TM | 3MYCA | 0.99534 | 97.70992 | 0.01924 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||||
| Lowest_TM | 2K6NA | 0.65695 | 95.52239 | 0.8452 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||
| 2RJWA | 2RJWA | 2RJWA | ||||||||||||||||||||||
| Highest_TM | 3MYAA | 0.99218 | 100 | 0.01924 | Y | Y | ||||||||||||||||||
| Lowest_TM | 2K6NA | 0.66794 | 95.52239 | 0.8452 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||||
| 2RJXA | 2RJXA | 2RJXA | ||||||||||||||||||||||
| Highest_TM | 2RJWA | 0.95017 | 97.01493 | 0.00192 | Y | Y | Y | |||||||||||||||||
| Lowest_TM | 2K6NA | 0.66111 | 91.04478 | 0.84712 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||
| 2RJYA | 2RJYA | 2RJYA | ||||||||||||||||||||||
| Highest_TM | 1YU5X | 0.9985 | 97.70992 | 0 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||||||
| Lowest_TM | 2K6NA | 0.67275 | 93.12977 | 0.84712 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| 3MYAA | 3MYAA | 3MYAA | ||||||||||||||||||||||
| Highest_TM | 2RJWA | 0.99218 | 100 | 0.01924 | Y | Y | ||||||||||||||||||
| Lowest_TM | 2K6NA | 0.67701 | 95.52239 | 0.82596 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |||||
| 3MYCA | 3MYCA | 3MYCA | ||||||||||||||||||||||
| Highest_TM | 2RJVA | 0.99534 | 97.70992 | 0.01924 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||||||
| Lowest_TM | 2K6NA | 0.66547 | 93.12977 | 0.82596 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | ||
| 3MYEX | 3MYEX | 3MYEX | ||||||||||||||||||||||
| Highest_TM | 3NKJA | 0.93842 | 98.43750 | 0.0197 | ||||||||||||||||||||
| Lowest_TM | 2K6NA | 0.63322 | 91.60305 | 0.84712 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | |
| Leu | Ala | Val | Ser | Thr | Pro | Ile | His | Lys | Asn | Asp | Glu | Gln | Tyr | Trp | Met | Phe | Gly | Arg | Cys | |||||
| Percent Change | 70.59 | 79.4 | 67.65 | 70.6 | 67.65 | 47.06 | 61.76 | 50 | 47.06 | 70.588 | 73.529 | 50 | 58.82 | 79.41 | 29.41 | 61.76 | 79.41 | 76.471 | 76.47 | 38.24 | ||||
Table 10: Overall change in TM_Score and amino acid residues |
||||||||||||||||||||||||




































