Open Access
Research Article
Max Screen >>
ISSN: 2454-4981
Copyright: © 2015 Ashok Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Human islet amyloid polypeptide (hIAPP), a 37 residue peptide hormone is an ingenious factor in pancreatic amyloid deposits found in cases with type-2 diabetes. Its aggregation into small toxic oligomeric species is presumed to be the reason for cells debilitation and demise in case of diabetic patients. In another case, rat islet amyloid polypeptide (rIAPP) dissents from hIAPP in only 6 residues, and accordingly, squanders its aggregation potential to form toxic oligomeric species. The atomic details of structure stability of toxic hIAPP and non-toxic rIAPP remain unclear. In the present study, in silico stability analysis were done on hIAPP and its oligomeric species along with 6 mutated structures and their oligomeric species which mimicked rIAPP. The stability analysis studies were done using molecular minimization, and implicit and explicit solvation techniques. The molecular minimization, implicit and explicit solvation techniques gave the structures which were stable and lead to aggregation forming toxic amyloids which results in dysfunction, and the unstable structures which formed non-toxic amyloid i.e. structures which lose aggregation ability and further not causing dysfunction. These non-toxic amyloids may act as therapeutic agents.
Keywords: hIAPP; Structure stability; Molecular minimization; Implicit solvation; Explicit solvation
Human islet amyloid polypeptide (hIAPP) is a 37 amino acid long hormone peptide with the following sequence "KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY" which is engendered by pancreatic islet β-cells and co-secreted with insulin in riposte to glucose [1]. hIAPP is adept indevising amyloid fibrils in vitro which are commonly soluble and natively unfolded in its monomeric state, but forms oligomeric amyloid in type 2 diabetes [2]. These amyloid deposits endowed in victims with type 2 diabetes are said to be its causative agent [3] and the main contributor to impeded insulin secretion and pancreatic β-cell death. Increasing evidence of impaired insulin secretion indicates that β-cell death are said to be allied to the interaction of hIAPP with the cellular membrane, which fastens peptide gathering [4], but the molecular structures of aggregated hIAPP remains elusive [5,2]. Isletamyloid is not itself the provocation of type 2 diabetes, but it leads to β-cell debilitation and cell death [6].
Rat IAPP, which dissents from hIAPP in only 6 of 37 residues with the following sequence "KCNTATCATQRLANFLVRSSNNLGPVLPPTNVGSNTY", where 6 mutations are shown in red font, which eludes its aggregation capability to form toxic amyloid species. Its monomeric and dimeric forms consist of Disulfide Bridge which also dissents from the hIAPP by 6 amino acids in the C-terminal region, but was still ineffectual to form fibrils [7-9]. The first exploratory facts on the isolated and unmodified dimers of hIAPP and non-amyloidogenic rIAPP revealed that the hIAPP dimers were more extended than those formed by rIAPP and likely cascades from extended monomers [10]. As the aggregation of hIAPP and rIAPP leads to toxicity and non-toxcity, respectively the analysis of structure stability needs to be done to get knowledge about stability of biological assemblies and their specific residues that differ in both hIAPP and rIAPP which are reasons for amyloid and non-amyloid formation.
A novel computational approach to the structural examination of ordered β-aggregation was exhibited and validated on amyloidogenic polypeptides. This procedure was based on the putrefaction of the sequence into overlapping protracts and equilibrium implicit solvent molecular dynamics (MD) simulations of an oligomeric system for each protract [11]. The structural stability of the in-register parallel aggregates sampled in the implicit solvent runs was further evaluated using explicit water simulations for a subset of the protracts [12-14] and these simulations showed that rIAPP forms compact helix and coil rich dimers, whereas hIAPP forms β-strand rich dimers that were generally more extended [15,16], which might be the reason of amyloidogenic fibrils formation.
In view of these facts, present study was designed to understand the atomic details of structure stability of wild type (similar to hIAPP) and mutated hIAPP (similar to rIAPP) models using minimization, implicit and explicit solvation approaches. These fields consist of theoretical and computational techniques used to model or mimic the behavior of hIAPP and its oligomers and rIAPP and its oligomers respectively.
The hIAPP crystal structure was retrieved followed by the construction of six point mutations and one multiple mutation on hIAPP structure. The biological assemblies of wild type model were compared with the predicted biological assemblies of mutation models to observe the respective effect on the structural stability of assemblies. These eight i.e. mutated (M (all mutations model), M1 (H-R mutation model), M2 (F-L mutation model), M3 (A-L mutation model), M4 (I-V mutation model), M5 (S-P mutation model), M6 (S-P mutation model)) and wild type (O) structures were analyzed for their structural stability by evaluating quality of oligomers using protein quality viewer. To study the molecular stability of the structures the energies of models were evaluated by molecular minimization and the effect of oligomerization of modeled protein was studied by predicted the biological assemblies. The explicit solvation of the models were studied and compared. Finally, implicit solvation using molecular dynamics approach which is a computer simulation of physical manoeuvre of atoms was done. The atoms were permitted to interact for a period of time (2.5ps), giving a view of the motion of the atoms. The trajectories of atoms were determined by numerically solving the Newton's equation of motion for a system of interacting particles, where forces between the atoms were defined by molecular mechanics force fields (AMBER force field). Finally, the obtained molecular data was statistically analyzed which helped in the comparative analysis between wild type and mutated hIAPP and conclude the differential effect of individual mutations. We also observed the effect of combinations of mutations where we discovered that they had drastic effect on the stability from monomer to polymeric form of hIAPP. Therefore, this study helped us in analyzing that why wild type hIAPP peptide shows toxicity and its mutated structures showed less or no toxicity at atomic structure level, and thus could be used as therapeutic agents in future.
The hIAPP sequence in FASTA format was retrieved from NCBI database and was made to undergo BLAST [17] search which provides fusion protein of hIAPP i.e. 3G7V (Islet Amyloid Polypeptide (IAPP or Amylin) fused to Maltose Binding Protein) whose crystallized structure was retrieved from PDB database [18]. It is known that six amino acid residue difference in rIAPP from hIAPP makes it loose its ability to form toxic amyloid species, therefore, these six amino acid residues were mutated in the sequence of 3G7V protein in the hIAPP region i.e. from residue 372-408 using Rosetta Backrub Server [19]. It has the feature of modeling point or multiple mutations by just specifying residue number and residue at which and with which mutation has to be done. In the wild type sequence Histidine (H) is mutated to Arginine(R) at 388th position, Phenylalanine (F) is mutated to Leucine (L) at 393rd position, Alanine (A) is mutated to Proline (P) at 395th position, Isoleucine (I) is mutated to Valine (V) at 396thposition, Serine(S) is mutated to Proline (P) at 398th position and Serine(S) is again mutated to Proline (P) at 399th position. The S to P mutations at 398th and 399th mutation are in accordance with the amino acid changes in rIAPP peptide sequence.
Further, these mutated sequences were compared to wild type hIAPP sequence by global pairwise sequence alignment using EMBOSS NEEDLE [20]. As the aggregation of full length hIAPP into small oligomeric species is growingly believed to be responsible for cell debilitation and demise whereas rIAPP loses its ability of aggregation therefore; the models were allowed to undergo oligomerization i.e. dimer and trimer formation respectively in order to study aggregation leading to toxicity in our models. The prediction of these biological oligomerized structures was done using Multimer Docking function of ClusPro server [21]. Then protein quality analysis of oligomers were done using protein quality viewer of Schrodinger Suite [22]. The potential energy of wild type and mutated models was calculated before and after energy minimization using Swiss PDB Viewer (SPDBV) [23] and Schrodinger Suite [22] in order to bring them to low minimum energy confirmation leading to the increase in the stability of structures. Then the explicit studies of the mutated residues was accessed using NetSurfP [24] which is a generic procedures for allocation of reliability scores applied to solvent accessibility prognostication and ABPS plugin in VMD to study electrostatic potential [25] and residue scanning was done using Schrodinger Suite [22]. Finally, the implicit studies were done using molecular dynamics simulations were calculated using MDWEB [26]. In this the MD simulations were done using NAMD force field for the total time period of 2.5ps with each time step of 1fs. The temperature was 300k and the output frequency was 50 steps i.e. 50 snapshots were observed. The trajectories obtained were analyzed using VMD. Same data was used to plot RMSD along trajectory, RMSD per residue and radius of gyration. The data at the end was statistically analyzed using clustering methods i.e. join tree clustering of STATISTICA software for all the used methods.
The hIAPP sequence in FASTA format was retrieved from NCBI and was made to undergo BLASTp search which searched fusion protein of hIAPP i.e. 3G7V as it had 41% query coverage with less than 0.1 e-value therefore it was selected as model for analysis, and its crystallized structure was retrieved from PDB database. 3G7V is Islet Amyloid Polypeptide (IAPP or Amylin) fused to Maltose Binding Protein whose monomeric structure was used for further analysis.
It is known that six amino acid residue differences in rIAPP from hIAPP evades its propensity to form toxic amyloid species, therefore, these six amino acid residues were mutated in the sequence of 3G7V and compared by global pairwise sequence alignment. Pairwise sequence alignment was done using Emboss Needle tool to observe the conserved regions between hIAPP and its mutated sequences. As the wild type sequence was compared with the all mutation containing sequences, it showed that H-R, F-L, A-P, S-P, S-P mutations have weakly similar properties and I-V mutation has strongly similar properties as indicated by dots (.) and colon (:), respectively. The global alignment includes all residues of both sequences as shown in Figure 1.
Protein structure prediction is the augury of the three-dimensional structure of a protein from its amino acid sequence. In order to study the impact of six residues difference of hIAPP from rIAPP, the 3G7V structure which corresponds to the hIAPP (wild type) was mutated using ROSETTA BACKRUB. The point mutation generates modeled structures and Rosetta scores for single and multiple point mutants in monomeric proteins shown in Figure 2. The 6 amino acid mutations lead to changes in the secondary structure of hIAPP peptides. The 3 mutations A to P at 395th position, S to P at 398th position and S to P at 399th position changes helices and beta sheets into turns due to the exceptional conformational rigidity of proline which effects the secondary structure of proteins by acting as a structural disruptor. The 2 mutations F to L at 393rd position and I to V at 396th position changes aromatic and long aliphatic chains into aliphatic amino acids leading to no interactions leading to disruption of secondary structural elements and either to the formation of loops. The mutation H to R at 388th position leads to decrease in basic character of amino acids again affecting the secondary structure of hIAPP. In the Figure 2, it is clear that the mutated structure loses its helical property and becomes loops, which may be the reason leading to loss of aggregation. The models with minimum Rosetta scores were used as mutated hIAPP models.
The aggregation of full length hIAPP into small oligomeric species is increasingly believed to be responsible for cell debilitation and demise whereas rIAPP loses its ability of aggregation. After structure prediction, the mutations showed differences from the wild type model, and these mutations may be responsible for the loss of aggregation. Thus, due to the above mentioned reasons there exits the need of oligomerization to study of stability of hIAPP oligomers but due to mutation the oligomerization stability could be affected so protein biological assemblies' studies are important. Therefore, in order to study this loss of aggregation, the models were allowed to undergo oligomerization into dimer and trimer structures using the FFT-based rigid docking correlation approach, which evaluates the energies of billions of docked conformations on a grid. The energy obtained by ClusPro describes the form of a correlation function, which respectively helps in studying the aggregation properties in our modeled structures. On the basis of monomers wild type model obtained from BLASTp and mutated structures obtained from ROSETTA BACKRUB, the biological forms were predicted and evaluated using ClusPro. Biological Assemblies of ClusPro and their corresponding energies were analyzed using STATISTICA clustering method that is joining tree clustering as shown in Figure 3. The impetus of this design is to join together objects into successively larger clusters, using Euclidean distance measure of similarity or distance. A typical result of this type of clustering is the hierarchical tree which showed that models O-M3 and O-M5 are closely related and rests all models are distantly related with model O-M2 being the most distant model. These tree representations are for monomer, dimer and trimer respectively.
The quality of oligomers obtained from ClusPro was obtained using Schrodinger Suite's protein quality viewer. It gave us values for Root mean Square bond deviations, angle deviations, backbone, side-chain, peptide plannar deviations, side-chain plannar deviation, torsion plannar deviations as shown in Figure 4. The Figure 4 gives Ramachandran Value which was minimum for all models in monomers i.e. 6, M6 in dimers and trimers i.e. 14 and 27, respectively. The RMSD values for bonds is 0.012 for all oligomerised models, and for angles were minimum for all models i.e. 1.139 in monomers, M2 i.e. 2.127 in dimers, M1 i.e. 2.18 in trimers. The peptide plannar deviation for monomer wasminimum for all models i.e. 3.454, dimers (4.552) for M and trimer (4.919) for O. The side-chain plannar deviation for monomer was minimum for all models i.e. 0.005, dimers (0.01) for O and trimer (0.01) for O. The torsion plannar deviation for monomer was minimum for all models i.e. 1.276, dimers (1.608) for M and trimer (1.526) for M.
The models generated above were minimized in order to bring them to low minimum energy conformation and analyzing there potential energy. The structural irregularities can be corrected by applying the energy minimization, which moves the model in such a way that overall conformation has the lowest energy potential. Molecular minimization was done using Schrodinger Suite. The potential energy increases as we move from monomers towards dimers and then trimers as shown in Table 1. The minimum energy for monomers, dimers and trimers is for M i.e. -3355.59, -6901.18 and -10930.8, respectively and it decreases as we moved from monomers to trimers.
To further study the effect of solvent environment on the mutated residues in the all mutation models solvent accessibility studies were done using NetSurfP. After the mutations when solvent accessibility of mutated models was studied they showed difference according to the mutations present in each model. After solvent accessibility electrostatic potential was observed using ABPS plugin in VMD. The data is in the form of PQR format which gives us the information about the effective charge and radius of the residues present in the structure. The PQR data was also analyzed using STATISTICA clustering method that is joining tree clustering as shown in Figure 5. This tree representation shows that the O is closely related to M4 and the M is closely related to M1, and these two are distantly related to each other in explicit solvation studies. Further, the mutated residues were also scanned using schrodinger suite as shown in Table (2a-d). The Table 2 demonstrates residue positions which were mutated and their stability analysis, solvent accessibility, rotatable bonds, hydropathy and prime energies. These analyses were minimum for histidine mutation to arginine. After all the above analysis the models were ready for molecular dynamics simulation. Simulations show that wild type models form (trimer) exhibit high structural stability with being the minimal seed in the solution whereas in the mutated models there is loss of overall stability.
Molecular dynamics simulations also known as implicit solvation has the edge to accurately reproduce kinetics of non-equilibrium properties such as diffusion or folding times. One of the most important rudimentary properties to analyze is whether the protein is stable and close to experimental structure. Molecular Dynamics by MDWEB is a personal workspace providing standard protocols to prepare structures, run standard molecular dynamics simulations and to scrutinize the trajectories. Protein structure study to mimic the effect of solvent in protein structure molecular dynamics studies were performed for 2.5ps with each time step of 1fs. The temperature was 300k and the output frequency was 50 (steps). 50 snapshots were observed. Same data was used to plot RMSD along trajectory, RMSD per residue and radius of gyration. The wild type model had 64095 atoms giving 2 frames and the all mutations model had 65235 atoms giving 10 frames.
It is a well-known fact that rIAPP differs from hIAPP in only 6 amino acids residues out of 37 residues, but rats/mice do not prosper diabetes-like prodromes even when rIAPP is over expressed [1]. It also loses aggregation ability to form toxic amyloid species. Therefore, here we considered molecular modeling approach to understand atomic details of models i.e. hIAPP (wild type model) and rIAPP (mutation models) [6]. These fields consist of theoretical and computational techniques used to model or mimic the behavior of macromolecules [1]. hIAPP is a 37 residue peptide, it cannot exist freely as it loses its structural form therefore its sequence was retrieved from BLASTp search with database search set as Protein Data Bank in which 3G7V which is IAPP fused to Maltose Binding Protein was selected as it has 41% similarity and less than 0.1 e-value. It also had its crystal structure present in PDB. Therefore hIAPP structure was retrieved from PDB. Then pair wise sequence alignment for global alignment using Emboss Needle to study conservation in the properties of mutated residues between wild type model and mutation models where I-V mutation showed conservation in strongly similar properties as compared to other models i.e. H-R, F-L, A-P, S-P and S-P which showed conservation in weakly similar properties. Therefore, the difference in functionalities of hIAPP and rIAPP may be the dependent on all the mutations other than I-V (Figure 1). The wild type model (O) was mutated in order to generate seven mutation models viz. all mutation models (M), H-R mutation model (M1), F-L mutation model (M2), A-P mutation model (M3), S-P mutation model(M5) and S-P mutation model (M6) using Rosetta Backrub (Figure 2). 20 models for each mutation models were generated out of which model with lowest energy were considered on the basis of energies generated by the server.
The structural stability of the amyloid fibrils was due to aggregation in these fibrils which lead to the formation of toxic species. Therefore, the biological assemblies (dimers and trimers) of models were predicted using ClusProserver [13]. 10 assemblies were generated out of which one for each model was selected on the basis of balanced score. These balanced scores were analyzed using joining tree representation from STATISTICA software which showed that biological assemblies of wild type model were closely related to M6 (Figure 3). Then the quality evalution of oligomers was done using Protein quality viewer of Schrodinger Suite which showed that wild type oligomers were stable as compared to all mutations oligomers resulting in amyloid fibrils of hIAPP. Molecular minimization force field from SPDBV and Schrodinger Suite was used to calculate the potential energy of models. There was increase in total energies as we moved from its monomer to trimer form. But within the each form the models did not show very drastic change with the maximum energy of -2634.84 for F-L mutation (M2) model and minimum energy of -3355.59 for all mutation model (M) (Table 1).
In the case of dimer, the maximum energy is again for wild type model (O) i.e. -6417.83 and minimum energy is -6901.18 for all mutation model (M). For trimer, the maximum energy is again for wild type model (O) i.e. -10233.9 and minimum energy is -10930.8 for all mutation model (M) (Table 1). For monomers, dimer and trimer again the maximum energy was for wild type model and minimum for all mutation models as expected. The explicit solvation accessibility of biomolecule was calculated using NetSurfP. The relative solvent accessibility was minimum for isoleucine and its mutation valine in all models. Thus we conclude that the above mentioned amino acid residues have minimum accessible surface area. The electrostatic potential was calculated for those amino acid residues which have significant role in the peptide sequence (Table 2) (Figure 4). All atoms implicit solvent MD simulations were performed to considerate the structural stability and dynamics of hIAPP and its respective mutation models [7]. F-L mutation model had minimum deviation in the RMSD values as compared to the wild type model (Figure 5). Thus, we could conclude from the above studies that H-R, F-L and I-V are the mutations which have similar properties to that of hIAPP and the A-P, S-P and S-P are the mutations which have specific properties in the rIAPP which makes the rIAPP less toxic by not allowing aggregation of peptide when we move from monomers to trimers. The wild type oligomers (hIAPP) are stable as compared to all mutations models (rIAPP) oligomers which are unstable. This work provides a molecular basis for improving our understanding of interactions governing the self-assembly of amyloidogenichIAPP peptides and non-amyloidogenicrIAPP peptides. Therefore rIAPP peptides could be used as therapeutic agents for diabetes 2 patients [27].
The authors acknowledge the financial assistance provided by University Research Fellowship by Panjab University, Chandigarh, India to Ms. Kanika Gupta (reference no. 1717-1743/ESTT-I). We would also like to acknowledge the support provided by the online servers, tools and softwares i.e. Rosetta Backrub Server (https://kortemmelab.ucsf.edu/backrub/cgi-bin/rosettaweb.py?query=index), ClusPro Server(https://cluspro.bu.edu/login.php), SwissPDBViewer (https://spdbv.vital-it.ch/), NetSurfP (https://www.cbs.dtu.dk/services/NetSurfP/), ABPS Plugin of VMD (https://www.ks.uiuc.edu/Research/vmd/plugins/apbsrun/), and MDWeb (https://mmb.irbbarcelona.org/MDWeb/).For this article, the authors apologize to any researcher whose work was not cited because of length constraints.

![]()

 |
| Figure 1: The comparison between wild type hIAPP and mutated hIAPP (Black arrows demonstrates weakly similar properties while the red arrow signifies strongly similar properties) |
 |
| Figure 2: a) The wild type hIAPP, the circuled region is where mutation takes place. b) Shows mutated hIAPP, the circular region is the mutated region and is distorted due to mutations |
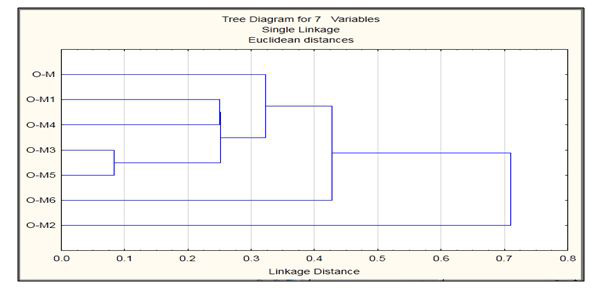 |
| Figure 3: Tree representations of monomers, dimers, trimers of hIAPP and it oligomers |
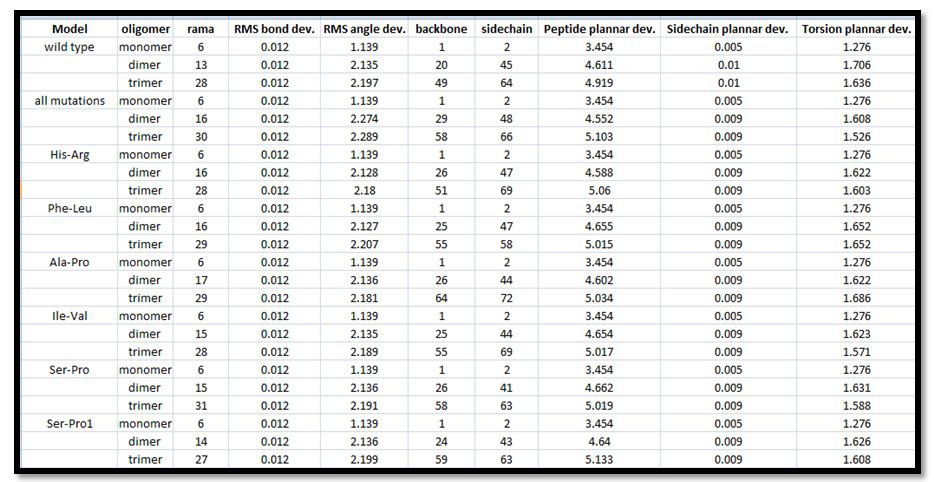 |
| Figure 4: Protein Quality View of oligomers for different models |
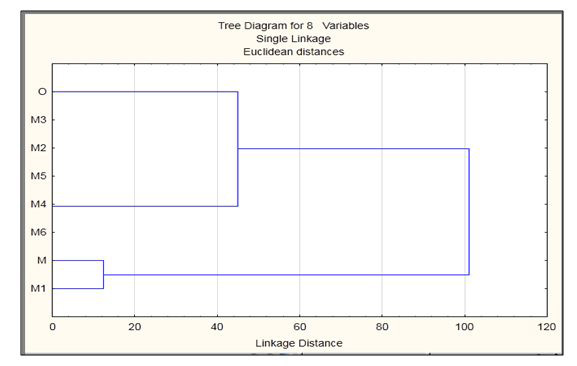 |
| Figure 5: The tree representation of Explicit Solvation |
| Models | Monomer Potential Energy | Dimers Potential Energy | Trimers Potential Energy |
| 0 | -2661.57 | -6417.83 | -10233.9 |
| M | -3355.59 | -6901.18 | -10930.8 |
| M1 | -2660.69 | -6441.33 | -10304.5 |
| M2 | -2634.84 | -6594.13 | -10265.2 |
| M3 | -2702.78 | -6588.19 | -10333.5 |
| M4 | -2652.18 | -6533.47 | -10266.9 |
| M5 | -2692.33 | -6591.74 | -10342.5 |
| M6 | -2697.95 | -6589.44 | -10361.7 |
| Table 1: The potential energies of monomers, dimers, trimers of hIAPP and its oligomers. The highlighted green shaded values show maximum energies for monomer, yellow shaded values show intermediate energies for dimer and cyan shaded values show minimum energies for trimers. The red shaded values are minimum energies within oligomers and it is for M model. The pink shaded values are maximum energies within oligomers and it for O and M2 model respectively | |||
| Residue | Original | Mutated | Stability (Solvated) | Potential Stability | SASA (total) |
| A: 388 | HIS | ARG | -17.41 | -157.23 | -12.29 |
| A: 393 | PHE | LEU | 4.91 | 0.43 | -9.41 |
| A: 395 | ALA | PRO | 48.62 | 60.68 | 7.01 |
| A: 396 | ILE | VAL | 7.28 | 4.97 | 0.08 |
| A: 398 | SER | PRO | 63.22 | 42.99 | 0.3 |
| A: 399 | SER | PRO | 38.05 | 29.86 | 4.76 |
| SASA (non-polar) | Prime Covalent | Prime SolvGB | Prime Solv SA | SASA (polar) |
| -10.79 | 2.37 | 29.23 | 0 | -1.5 |
| -6.14 | 3.44 | -0.09 | 0 | -3.27 |
| 26.33 | 53.37 | 1.14 | 0 | -13.48 |
| 0.08 | -5.12 | -1.34 | 0 | 0 |
| 101.75 | 56.55 | 4.78 | 0 | -99.67 |
| 49.48 | 36.18 | 17.99 | 0 | -44.71 |
| Total Rotable Bonds | Pime Coulomb | Hydropathy | Prime Lipo |
| 2 | -63.52 | -0.08 | -7.28 |
| 0 | -0.46 | 0.02 | 4.47 |
| 0 | 28.26 | -0.54 | -2.28 |
| -1 | -0.12 | 0 | 5.43 |
| -2 | 15.48 | -0.42 | -3.4 |
| -2 | 9.01 | -0.18 | -6.18 |
| Prime vdW | Prime Energy |
| 2.64 | -37.58 |
| 1.64 | 9.1 |
| 2.03 | 82.66 |
| 1.64 | 0.49 |
| 20.68 | 96.78 |
| 8.4 | 69.96 |
| Table 2: Residue scanning for each mutation in hIAPP | |




































