Open Access
Review Article
Max Screen >>
ISSN: 2393-9060
Copyright: © 2016 Giraudo S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
The rise of chronic non-communicable diseases (CNCD), mainly cardiovascular disease, obesity and cancer, has been assumed to be a consequence of the increased intake of fatty foods, especially saturated fats and cholesterol. This assumption triggered a body of guidelines, recommending a decrease in fat and an increase in complex carbohydrates, with the use of some oils that are thought to be beneficial for health. However, these recommendations have not produced the expected results. Not only has CNCD not decreased, but instead it has increased markedly. In this paper we discuss the possible mechanisms for this unexpected outcome. Our main hypothesis is that an increase of the anabolic–inflammatory stimulation of the circulatory system and other tissues may contribute to CNCD. Hence, it would be prudent to rethink nutritional advice for at risk populations.
Keywords: Diet and chronic non-communicable diseases; Cardiovascular disease; Saturated fats and health; Obesity and fat; Diet and cancer; Cancer growth factors and insulin-resistance and diet
In the last hundred years, a major part of the developed world has shifted from a predominance of diseases caused by nutritional deficit to diseases caused by nutritional excess. Increasing income, improvement in the production and consumption of food, and an increase in the availability and intake of palatable processed food has triggered an epidemic of chronic disease.
Cardiovascular diseases and cancer are the two main causes of death in developed countries. In 2014, with over a total of 56 million deaths worldwide, 68% were caused by chronic non-communicable diseases (CNCD): 31% of them by cardiovascular diseases (17.5 million), 14.6% by cancer (8.2 million), 2.7% by diabetes (1.5 million) and 7.1% by respiratory diseases (4 million). These four groups of diseases account for 82% of all non-communicable diseases [1].
Taking into account that in the last 10,000 years the spontaneous mutation of nuclear DNA was calculated to be only of 0.0005%, it can be concluded that the increase in the rate of degenerative illness (such as atherosclerosis, tumors or fatty liver) is certainly not of a genetic origin but mainly attributable to environmental changes, in particular to eating habits [2,3].
The main dietary recommendations for populations have been aimed at controlling cardiovascular diseases and obesity. These suggestions were inspired by the hypotheses that cholesterol and saturated fats were responsible for injuring arterial walls, and also that obesity was caused by an energetic imbalance due to an excess of dietary fat and inadequate physical activity [4]. These dietary recommendations encouraged a decrease in the consumption of animal products (saturated fats, cholesterol and total fats) in spite of the fact that more than 50% of the calories ingested by many populations usually come from sources other than animal products [5-7].
These official guidelines of the last 20 years have encouraged food producers and the pharmaceutical industry to develop products aimed at decreasing both dietary fat and cholesterol with the hope that this would in turn contribute to decreasing the levels of these substances in circulating blood. As a result of these recommendations by the USDA, the American Heart Association (AHA) and the American Diabetes Association (ADA), the intake of saturated fat has decreased from 13.5 to 10.6% of daily calories for males and from 13.0% to 11.0% for women, while the consumption of carbohydrates has increased from 42.4% to 49.0% for males and 45.4% to 51.6% for women [8]. From 1970 to 2008, calorie intake has increased 616 Kcal due in large part to added fats (+213 Kcal) and flour and grain products (+193 Kcal) (Table 1).
Data from 10 cohort studies and the meta-analysis of 33 randomized and controlled trials (73,589 people) in developed countries showed that low fat diets were associated with a slightly lower body weight [10]. However, the reported difference of 1.6 kg (on average), although statistically significant, does not correlate with the increase in BMI (from 25 to 30) [10].
In China, an increase of 10% in fat calories showed a non-significant BMI increase of only 0.1 in adolescents and of 0.003 in adults. In South Africa, a rather high incidence of overweight (>55%) is related to a low fat consumption of 22% [11].
On the other hand, it is important to bear in mind that in the US, a decrease of fat consumption from 37% to 33% in men and from 36% to 33% in women has resulted in an increase of overweight and obesity in the past 40 years [12,13]. Obesity increased from 12.6% in 1991 to 33.9% in 2015 while overweight increased from 36.7% in 1995 to 66.9% in 2010 as reported by the World Health Organization [14]. In Argentina, the prevalence of obesity has increased from 14.6% to 18.0% from 2005 to 2009 and overweight from 34.4% to 35.4% [15].
Obesity is a result of energy imbalance which may be influenced by many elements, including non-caloric factors that might be influencing body composition. These may include 1) the feeding cycle (e.g. in animal experimentation, a reversed light cycle produces a greater increase in weight with the same energy consumption); 2) metabolic regulation (e.g. an impairment of the adipose insulin receptor protects against the weight increase observed in controls); 3) changes in the muscular efficiency after weight loss; 4) the influence of non-energetic nutrients ( e.g. deficits of calcium or vitamin D) which decrease the loss of weight produced by low caloric diets; 5) biphenyls; and 6) intestinal microflora, whose composition is related to the level of adiposity [16-26].
Although neither the Food and Agriculture Organization (FAO) nor the World Health Organization (WHO) consider dietary fat as particularly causative factor in obesity or atherosclerosis; the Institute of Medicine-National Academy of Science (IOM-NAS), American Heart Association (AHA) and Academy of Nutrition and Dietetics (AND) recommend a maximum of 35% for dietary fat intake and maximum of 35% protein, while allowing up to 50 to 65% of carbohydrates. Thus for a diet of 2000 Kcal, about 300 g of glucose (~ 60%) would be recommended [5]. An intake of 300 g of glucose appears to be too much if we consider that: 1) the human storage capacity is about 320g (see table 2), 2) the daily estimated requirement is about 60-65 g of glucose 3) the difficulty of eating this amount of glucose if one only consumes natural foods and beverages [27] (Table 2).
It should also be noted that concentrated carbohydrates, often associated with fats and present in processed foods, are a main source of solid fats and added sugar (SoFAS) in the diet. The USDA Guidelines for 2015 suggest that intake of SoFAS and products obtained by refining grains typically exceed the recommended levels [29]. The main five sources of SoFAS for the population over 2 years of age are shown in table 3:
Solid fats are found in coconut oil, palm oil, butter, tallow, bacon, shortening and solid margarine (these last two contain partially hydrogenated oil which contains trans-fatty-acids). In liquid form, saturated fat is also found in olive oil, soy oil, corn oil and sunflower oil [30] (Table 4).
An association between high-fat diets and impaired insulin action has been observed in numerous in vivo and in vitro studies. Studies suggest that an intracellular excess of fatty acids may result in a decrease in insulin sensitivity affecting the beta cells of the pancreas [31,32]. This decrease in insulin sensitivity results in an increase in the release of fatty acids from adipose tissue, which are then deposited in other tissues, preventing the oxidation of glucose. As a result of this glucose sparing, the circulating level of glucose increases, producing glucotoxicity [32].
Hence, excess fatty acids may induce not only insulin resistance, but also glucotoxicity. In contrast, energy restriction improves insulin sensitivity, reduces fasting glucose, and insulin concentration. Thus, energetic restriction does not cause gluco- or lipotoxicity, on the contrary, it rather corrects it. In addition, restriction does not promote morbidity or mortality, and instead, it tends to extend life [33].
These beneficial actions of energy restriction may be attributed to an increase of transcription factor FOXO1, which is normally restrained by insulin, and to a lesser activity of growth factor1 (IGF-1) and of mTOR (mammalian target of rapamycin), both of which are stimulated by insulin [34,35].
The respiratory quotient produced by a Western diet is approximately 0.80-0.85, which is equivalent to the oxidation of approximately equal amounts of glucose and fatty acids. Indeed this oxidation profile occurs with formulations of calories from 41% fat, 45% carbohydrate and 14% protein. A similar oxidation level is observed with whole milk which has a profile of 53% fat, 41% carbohydrate and 8% protein profile [36]. In contrast, most of foodstuff ingested by indigenous populations is characterized by low carbohydrate, and contains a high percent of fat and protein. For example, Massai warriors whose diet is mainly meat, milk and blood and is estimated to include 60% of fat calories (33% saturated fat), exhibit no cardiovascular disease, despite their low level of physical activity [37-40]. Inuit Eskimos have an intake of more than 70% of fat calories, but chronic diseases are uncommon among them [41]. Tokelau aborigines, native of a New Zealand territory island, obtain 53% of their daily energy intake from coconut oil (which is 90% saturated fat), 13% protein and 34% carbohydrate. This notwithstanding, the prevalence of cardiovascular diseases is rather low in this population [42]. The Mediterranean diet, which contains 45% of fat, is highly recommended for diabetic patients because of its favorable effects on blood glucose and the prevention of cardiovascular diseases [43]. The Atkins diet, in which more than 50% of the energy comes from fat-mainly saturated ones- shows a better metabolic profile than the AHA or the very low fat Ornish diet [44].
It is also important to bear in mind that meta-analysis of diet composition proves no clear relationship between saturated fat intake and cardiovascular disease [45]. In addition, coronary disease risk cannot be predicted by reference to the level of saturated fat in meals [46].
However, eleven prospective studies of American and European cohorts have shown that replacing saturated fats by carbohydrates increased coronary risk [47]. In the famous “Study of the Seven Countries” by Ancel Keyes, several of the populations analyzed did not show a correlation between saturated fat intake and coronary mortality [48]. Further, the male populations of Finland, USA, Zutphen (Netherlands) and Crete differ substantially in coronary mortality in spite of the fact that they have a similar daily fat intake of approximately 40% [49]. In contrast, as seen in Figures 1 and 2, seven European countries with lower intakes of saturated fat (under 7.5%) had the greatest rates of mortality due to heart disease, whereas seven countries with the highest intakes (around 15%) had the lowest rates (Figures 1 and 2) [49].
In addition, a review of 146 prospective papers and 43 randomized clinical reports shows that cardiovascular diseases were not attributable to fat intake [50,51]. In a 2010 paper, “Fat and Fatty Acids in Human Nutrition”, FAO-OMS acknowledge that the evidence relating saturated fat intake to metabolic syndrome and cancer is insufficient [52]. This conclusion is supported by the fact that about 50% of the fatty acids in cell membranes are saturated and their proportions are almost indifferent to any changes in diet. This phenomenon also occurs with mono-unsaturated fats [53]. Conversely, essential fatty acids in the membrane are quickly influenced by the quality of fats ingested [53].
The daily fluctuations in blood insulin levels together with changes in insulin receptor sensitivity suggest that cellular glucose consumption is regulated, especially for the tissues which are glucose dependent [54]. The variables regulating the entrance of glucose to the cell are glucose transporter 4 (GLUT4), the accumulation of certain lipids, and the effect of the ribosomal protein S6K (activated by mTOR), among others [55-58]. The fluctuations of the hormonal and tissue metabolites of this system prevent elevated glucose entry to the tissues and results in excessive postprandial hyperglycemia, thereby protecting those tissues which do not regulate glucose entry [59].
Non-enzymatic glycosylation of tissue proteins, imperfect electron transport in the respiratory chain and increase in the hexosamine pathway lead to an excessive production of reactive oxygen species (ROS), which are a direct cause of the glucotoxic injury [60-63]. ROS, as sensors of oxidative stress contribute to the impairment of insulin-mediated glucose uptake thereby preventing local glucotoxicity at the expense of raising glycemia and insulinemia [64,65]. This increased glycemia and insulinemia in turn leads to greater glucose utilization by: a) increasing glucose oxidation by reducing the adipose tissue release of fatty acids and b) stimulating glucose transformation in fat and cholesterol via the “de novo” synthesis of fatty acids and cholesterol [66-71]. Hyperglycemia and hypertriglyceridemia may result from the above process which may be considered as signs of calorie excess, due to cellular impairment of glucose and triglyceride utilization [72].
Due to the fact that an abnormal increase of insulin resistance is found in almost 30% of normal weight individuals the progressive increase of weight during aging could in part be attributable to excess circulating nutrients and to hyperinsulinism (characteristics of metabolic syndrome), due to the insulin resistance [73]. Thus, excess body weight or hyperphagia may be the result, rather than the cause of the hyperinsulinemia and excess circulating nutrients.
In support of this hypothesis, it has been shown that after experimental injury of the hypothalamus, there is an increase of insulinemia that precedes the hyperphagia and obesity Ransom [74]. Hyperinsulinemia and a greater trophism of intestinal mucous preceded the development of obesity after ventromedial hypothalamus injury [75]. Likewise, the deletion of the insulin gen 2 (Ins 2 -/-) in mice who continue to exhibit some insulin activity due to intact activity of gen insulin 1(Ins 1 -/+), showed no hyperinsulinemia, obesity or metabolic syndrome when exposed to obesogenic diet. It was suggested that their fat cells were reprogrammed to waste the excess nutrients as heat, CO2 and water [76].
In 1464, Luigi Cornaro proposed the benefits of restricted eating in “Discourses on the Temperate Life” [77]. Nematodes, insects and mammals respond to dietary restriction by altering their glucose homeostasis and insulin signaling. It has been suggested, that limiting calories can modulate inflammation, insulin sensitivity, cellular cycle, tissue proliferation and apoptosis [78].
By increasing bioavailability of IGF-1 insulin mediates the anabolism and cellular proliferation associated with overeating. This increase in IGF-1 in turn coincides with the over- expression of the insulin receptor and its increased sensitivity in malignant cells [79,80].
m-TOR, a protein kinase, is an integrator of metabolic (ATP/AMP, AMPK), nutritional (glucose, amino acids, especially leucine) and hormonal signals (insulin and IGF-1) all related to cell proliferation and insulin resistance [81]. AMP-activated protein kinase (AMPK) promotes activity of Forkhead box protein 01 (FOXO1) which is inhibited by insulin and which has action against stress, tumors and inflammation. mTOR activation releases ribosomal protein S6K1 which increases insulin resistance and regulates cell proliferation and apoptosis [82-88].
The Akt-mTOR pathway remains sensitive to insulin in type 2 diabetics, obese non-diabetics and insulin-resistant obese non-diabetics and is involved in atherogenesis by increasing collagen, cellularity and pro-inflammatory cytokines in vascular tissues [89-91].
Reduction of circulating insulin levels caused by food restriction contributes to decreasing m-TOR activity which may improve some complications of diabetes, such as nephropathy, retinopathy, atherosclerosis and coronary diseases [92-95] (Figure 3).
AMPK: (AMP activated protein kinase): indicates energy associated with ATP/AMP level
BAD: (Bcl-2-associated death promoter): Protein of apoptotic family such as a Bcl-2
FOXO: (transcription factor forkhead box). Has anti-stress, anti-proliferative, anti-inflammatory and hyperglycemic properties
GH: (Growth Hormone): over-regulate IGF-1 IGF-1: activate PI3K and MAPK
AKt: Serine-threonine protein kinase or protein kinase B
S6K: ribosomal S6 protein kinase: is required for protein synthesis and promotes insulin-resistance
m-TOR: active S6K, sensor of amino-acids and energy
IGF-1: Insulin-like growth factor 1
IRS - PI3K: Insulin activation of phosphatidylinositol 3-kinase
GLUT4: Glucose transporter type 4
MAPK: Mitogen-activated protein kinases
An apparent paradox exists such that the reduction of insulin signaling via IGF-1 increases life expectancy. But resistance to insulin, which also reduces insulin signaling/IGF-1, leads to decreased life expectancy and to diabetes. In the first condition, the low insulinemia produces low mTOR activation, while in the second case, insulin resistance, mTOR activation is enhanced [96,97].
The Western diet is rich in processed sugars (such as fructose) and fat, has been associated with metabolic impairment in nonhuman primates and hepatic steatosis in obese rats [98,99]. Excessive intake of fructose has been linked with weight gain, diabetes, inflammation, hyperuricemia, hypertension, vascular diseases and fatty liver. Even when consumed in low amounts and for a short period of time (3 weeks), the consumption of soft drinks leads to a reduction of the size of LDL, changes in glycemia and an increase in C-reactive protein [100]. These changes help explain the association between fructose and vascular diseases and/or metabolic syndrome. Hepatic metabolism of fructose causes fat accumulation and inflammation in the liver. Non-alcoholic fatty liver disease may result in steatohepatitis fibrosis (20-50%), cirrhosis (20%) or hepatocellular carcinoma (1-5%) [101-104].
High fructose corn syrup became a market product in 1970 and between 1970 to 2000, its consumption increased from 0.5 lb per capita in 1970 to 55.3 lb per capita in 1993 [105]. Fructose consumption increased from 37 g/day in 1977-78 to 55 g/day in 1988-94 (NHANES III) [106]. In US, the average consumption of added sugars and sweeteners in 2010 was 19% higher than in 1970 [107].
High fructose corn syrup is a mixture of different proportions of glucose and fructose, but it typically contains 55% of fructose. It has replaced sucrose as sweetener in most soft drinks, candies, canned fruits, ice cream, cookies, etc [108]. After its duodeno- jejunal absorption, fructose arrives via portal vein at the liver, where it is extracted in high amounts as its entrance transporter is not regulated. In addition, the fructose transporter is not present at either the brain or the pancreas, thus fructose does not trigger central satiety signals or insulin secretion in proportion to the amount ingested [109-112]. When fructose enters the liver it is phosphorylated and transformed in fructose-1-phosphate (F1P) [113]. Chronic consumption of fructose promotes a quick depletion of ATP [114]. F1P is a precursor of trioses which enter the “de novo” synthesis of triglycerides and cholesterol [113].
An acute cellular deficit of ATP activates the adenosin-monophosphate-deaminase-1, which in turn, increases the synthesis of uric acid thereby increasing the risk of gout and hypertension (due to the uric acid inhibition of endothelial synthetase of nitric oxide) [115-119]. These disorders have been observed in adolescents consuming high amounts of soft drinks. Hypertension combined with hyperuricemia may promote vascular damage, even when treated with antihypertensive drugs [120].
In mice, fructose increases the translocation of intestinal bacterial toxins. This translocation causes an increase of TNF alpha, resulting in hepatic steatosis [121].
If the fructose stream is substantial, and/or if glycogen deposits are full, the acetyl CoA produced by fructose can be a substrate for “de novo” lipogenesis, which can increase up to 10-fold [122,123]. Fatty acids can be exported as low density lipoproteins, resulting in hypertriglyceridemia (see table 5) and/or can be deposited as fat in the hepatocyte [124,125].
The effect of carbohydrates on responding elements such as ChREBP, is higher for fructose than for other sugars, as isocaloric replacement of glucose by fructose increases intrahepatic fat by 38% in 8 days [126-129] (Table 5).
After 8 weeks of ingesting 25% of calories from soft drinks containing fructose, subjects showed a greater weight gain (+1.4 kg) and a significant increase of intra-abdominal fat fructose as compared to glucose ingestion [124]. Thus, in hypercaloric conditions, fructose may produce “de novo” lipogenesis, hypertriglyceridemia, liver steatosis, insulin-resistance and increased ROS production, with risk of hepatocellular damage [129]. Taking into account its lipogenic potential, its inflammatory action, and its triggering of insulin-resistance, fructose may be considered as an etiopathogenic factor of NAFLD and cardiovascular risks, such as dylipidemia, hyperuricemia, hypertension and obesity.
In the last 100 years, in the developing world, diseases due to nutritional deficit have been replaced by diseases due to nutritional excess. These changes parallel an increase in the production and consumption of highly processed foods. As a consequence, health authorities have promoted a diet to decrease cardiovascular disease and obesity, namely a diet consisting of low-fat foods in conjunction with high levels of “healthy” carbohydrates (45-65% of calories), such as whole grains, fruits and vegetables.
Despite these recommendations, there has been limited success with this strategy. While deaths attributable to heart disease have declined in the last 20 years, obesity rates continue to climb. Of course, a sedentary lifestyle is a contributor to chronic disease. Genetics also plays a role, but most likely is not an immediate cause of this increase as our genetic pool cannot change in such a relatively short amount of time.
Perhaps it is time to re-visit the dietary prescriptions advanced by government organizations and health associations. Indeed, studies have indicated that dietary macronutrient composition can vary widely with no effect on body weight, as long as energy intake is balanced by output [130]. Due to the relatively satiating effects of protein and fat, it may be prudent to promote higher intakes of these macronutrients and lower intakes of carbohydrates.

![]()

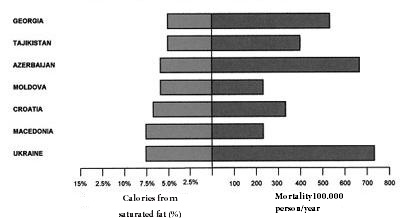 |
Figure 1: Mortality caused by cardiovascular decease and low intake (≤7.5 %) of saturated fat |
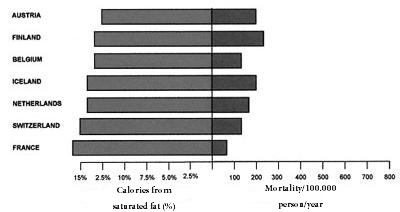 |
Figure 2: Mortality caused by cardiovascular decease and high intake (>7.5%) of saturated fat |
 |
Figure 3: Main signalization ways activated by insulin receptor and growth factors (taken from references modified) [79-82] |
FOOD INTAKE CHANGES BETWEEN 1970 AND 2008 (expressed in calories) |
||||
|---|---|---|---|---|
Year |
1970 |
1990 |
2008 |
1970 vs 2008 |
Components |
Calories |
|||
Dairy |
155 |
260 |
257 |
+102 (+66%) |
Fruits |
71 |
85 |
87 |
+16 (+23) |
Vegetables |
125 |
126 |
122 |
-3 (-2%) |
Meat - eggs and nuts |
463 |
453 |
482 |
+19 (+4%) |
Flour and grain products |
432 |
573 |
625 |
+193 (+45%) |
Caloric sweeteners |
402 |
446 |
459 |
+57 (+14%) |
Added fats and oils |
403 |
446 |
616 |
+213 (+53%) |
Other dairy fats |
6 |
15 |
25 |
+19 (+417%) |
TOTAL |
2057 |
2404 |
2673 |
+616 (+30%) |
Table 1: Food Intake Changes Between 1970 AND 2008 (expressed in calories) [9] |
||||
CARBOHYDRATES IN 70 KG ADULTS IN POST-ABSORTIVE STATE (adapted from reference) [28] |
|||
Tissue mass |
Liver (1.800 kg) |
Muscle (35 kg) |
Extra-cellular fluids (every 10 liters) |
Glycogen and glucose and tissue concentration |
72 g (4%) |
245 g (0.7%) |
10 g (0.1%) |
Table 2: Carbohydrates in 70 Kg Adults in Post-Absortive State (adapted from reference) |
|||
SOLID FATS (SoF) |
% CONTRIBUTION IN FOODS |
ADDED SUGARS (AS) |
% CONTRIBUTION IN FOODS |
Rice, pasta, grain dishes |
5 |
Soft drinks, energy drinks |
28 |
Pizza |
6 |
Grains |
8 |
Dairy |
13 |
Fruit drinks |
11 |
Burgers, sandwiches, meat, poultry, seafood dishes |
23 |
Dairy desserts |
4 |
Vegetables |
7 |
Candy |
31 |
TOTAL |
54 |
82 |
|
| Table 3: Five sources of SoFAS for the population over 2 years of age [29] | |||
% OF FATTY ACIDS |
SOY OIL |
OLIVE OIL |
CORN OIL |
SUNFLOWER OIL |
Saturated |
14.0 |
14.0 |
17.0 |
12 |
18:1 (n-9) |
25.0 |
72.0 |
30.0 |
33 |
18:2 (n-6) |
52.0 |
11.0 |
50.0 |
52 |
18:3 (n-3) |
7.0 |
1.0 |
2.0 |
Trace |
n-6 / n-3 ratio |
7.42 / 1 |
11 / 1 |
25 / 1 |
52 / 1 |
Table 4 (slightly modified) from reference [30] |
||||
IMPACT OF FIVE DIFFERENT CARBOHYDRATES ON CIRCULATING TRIGLYCERIDS AFTER A MEAL CONTAINING 40 G. OF FAT* |
||
Treatment |
Lipidemia post-prandial total (mmol.L-1. 7 h-1) |
Maximal increment of postprandial triglycerides (mmol/L) |
50 g fructose |
4.23 ± 2.10a |
1.06 ± 0.52a |
100g sucrose |
3.77 ± 1.90a |
0.96± 0.46a |
50 g glucose (iv) |
3.55 ± 1.68b |
0.86±0.44a |
50 g sucrose |
2.75 ± 1.33b |
0.71±0.32c |
Meal |
2.42 ± 1.24b |
0.63±0.33c |
50 g glucose (oral) |
2.11±1.23c |
0.57±0.27c |
*Media ± SD. n=21 (9 males, 12 females) |
||




































