Open Access
Review Article
Max Screen >>
ISSN: 2455-7641
Copyright: © 2016 AlDallal S. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Thalassemia is one of the most common genetic disorders worldwide and presents a major public health problem and social challenge in parts where the frequency is high. The symptoms of the disorder are modulated by various environmental, racial and genetic factors. Therefore, dental specialists are obligated to have knowledge towards the nature of the disorder and its effect on dental health. Cooperation with a hematologist is recommended in every dental treatment. This article discusses the different types of thalassemia and their orthodontic manifestations in patients and presents recommended techniques for avoiding any possible problems.
Keywords: Thalassemia; Dental care; Oral complications
List of Abbreviations: HbH: Hemoglobin H; RBCs: Red blood cells; HbF: Fetal hemoglobin
Hemoglobinopathies, mainly sickle cell anemia and thalassemia, are genetic blood disorders that are globally widespread, and about 5% of the world’s population carry the genes responsible for hemoglobinopathy [1]. Thalassemia is a group of congenital disorders characterized by a deficient synthesis of both alpha (α) and beta (β) chains of hemoglobin. Depending on the globin chain, which has quantitative synthesis defects, thalassemia can be divided into two major groups, i.e., α-thalassemia and β-thalassemia [2].
The consequences of these genetic disorders include anemia characterized by the presence of hypochromic and microcytic erythrocytes. The most commonly observed variant is the so-called thalassemia trait, which is associated with mild and insignificant anemia. On the other hand, the more serious forms of β-thalassemia are infrequently observed in patients (Table 1).
The disturbances in globin chain production can lead to infections due to encapsulated bacteria in thalassemia patients, which can be fatal at times. Severe anaemia, iron overload, splenectomy, and a range of other immune abnormalities can be considered as some of the risk factors causing infections in these patients [3,4]. Infections are considered as the second most common cause of morbidity in thalassemia patients [4].
Early detection of thalassemia can aid in opting for different treatment measures, such as blood and bone marrow transfusions, iron chelation, and folic acid supplements. Red blood cell transfusion is now-a-days considered as a prime treatment option in patients having both moderate and severe thalassemia [5]. These clinical procedures can eventually help in preventing the occurrences of infection in thalassemia patients.
Molecular studies have detected that the loss of α-gene function is related to either gene deletion or mutations, which cause terminator codon mutations and hypofunctional genes responsible for various α-thalassemia syndromes [6]. α-thalassemia can be suspected based on factors such as a family history of anemia and ethnic and geographic backgrounds, particularly if the patients are from the Middle East, Southeast Asia and North Africa, where the disease is highly prevalent. The diagnosis is suspected in patients showing microcytic hypochromic anemia (not caused by iron deficiency) with normal HbA2 levels in Hb electrophoresis. The α- or β-thalassemia traits, also known as silent carriers, are found in heterozygous individuals with impaired production of α- or β-globin chain depending on the gene involved. The syndrome does not generate any clinical signs (asymptomatic), i.e., it may be present with either normal blood count or morphology or with mild microcytic hypochromic anemia. The presence of an enlarged spleen (splenomegaly) is also rare in such cases. A differential diagnosis must be made to distinguish patients with α-thalassemia from those with iron deficiency anemia. No specific treatment is required for patients with silent carriers unless the patient is anemic [7].
Patients with hemoglobin H (HbH) disease present moderately severe deficiency in the production of α-globin chain. These patients have mild to moderate microcytic hypochromic anemia with hemoglobin (Hb) levels of 8–10 g/dL. Moreover, the physical examination of these patients shows hepatosplenomegaly. Exacerbation of anemia can be induced by acute infections, exposure to oxidative stress, folic acid deficiency, and pregnancy. These can be treated by folic acid supplementation and periodic blood transfusions when required [8]. Fetal hydropexis is an intrauterine manifestation of the severe form of α-thalassemia [9].
β-thalassemia is caused by chromosome 11 mutation that may affect the β-globin chain production. The disease is characterized by a severe hemolytic anemia, and it is the most single gene abnormality. It has over 200 mutations, and most of them are rarely observed. β-thalassemia is found in patients with intense alteration of β-globin chain production. In these patients, the α-chain production is normal; however, the lack of β-chains for binding results in an accumulation of α-chains. The α-chains are insoluble, and thus, tend to precipitate, forming intracellular inclusions that deform the structure of the red blood cells (RBCs), resulting in its premature destruction within the bone marrow and spleen. This further result in an ineffective erythropoiesis, which triggers an increase in erythropoietin levels with a rise in the output of erythroid cells and erythroblasts associated with bone expansion, resulting in typical facial deformities [10,11]. About 20 common alleles comprise 80% of the known thalassemia cases worldwide, and around 3% of the world populations carry genes of β thalassemia [12]. Iron deposition due to repeated blood transfusions causes many β-thalassemia complications. Moreover, the excess accumulation of iron in different tissues damages organs, mainly endocrine glands, liver and heart [13]. The most prominent endocrine complication is the failure of normal development and growth retardation [14].
Based on the need of regular blood transfusions, β-thalassemia patients can be categorized as thalassemia major and thalassemia intermedia. Thalassemia major patients need regular blood transfusions to survive. Such patients also experience bone deformations due to marrow expansion within the skull. On the other hand, patients with thalassemia intermedia do not need regular blood transfusions [15].
In β-thalassemia minor (also known as β-thalassemia trait), the clinical manifestations are usually mild, and the patients with this syndrome usually have a good quality of life. Although anemia in these patients is clinically insignificant and does not require any specific treatment, some patients have occasionally been reported with mild bone changes, splenomegaly, cholelithiasis, and leg ulcers. As the diagnosis of β-thalassemia minor is tricky, one must rule out the existence of iron deficiency anemia that may alter the elevated HbA2 levels. Depending on the underlying genetic mutation, high levels of fetal hemoglobin (HbF) are also seen. The RBCs in β-thalassemia are microcytic hypochromic in nature with a mean corpuscular volume <79 fL [16].
Patients with thalassemia intermedia have a moderate anemia and show a heterogeneous hematological picture ranging in severity from β-thalassemia minor to β-thalassemia major [17]. The need of transfusions differentiates β-thalassemia intermedia from β-thalassemia major [16].
β-thalassemia major, also known as Cooley anemia, denotes compound heterozygous or homozygous forms of the disease, which is characterized by severe anemia, massive ineffective erythropoiesis and hemolysis [18]. Clinical manifestations of patients with β-thalassemia major appear in infancy and include severe anemia, jaundice, extreme pallor or failure to thrive, decreased physical activity, poor feeding, irritability or/and increased somnolence. Furthermore, these patients are diagnosed with hepatosplenomegaly and frontal bossing with early signs of abnormal facies [19].
The orofacial manifestations of thalassemia are due to the bony changes, called Cooley facies, caused by ineffective erythropoiesis along with the development of bone-expanding erythroid masses (Figure 1). Occlusal abnormalities and bimaxillary protrusions are frequent in patients with thalassemia major. Facial and dental abnormalities include marked opened bite, poor spacing of teeth, saddle nose, and prominent malar bones. The skeletal changes result in upper lip retraction giving the child a “chipmunk facies” [20,21]. Furthermore, oral mucosa is pale most of the times [22,23]. As the condition is asymptomatic in α-thalassemia patients, mild anemia is expressed at the oral level by mucosal pallor [23]. Over-development of the maxilla frequently results in an increased over jet and spacing of maxillary teeth and other degrees of malocclusion (Figure 2) [24].
The patients with β-thalassemia major are mostly at risk of experiencing oral and facial problems due to bone marrow hyperplasia [25]. The radiographic alterations in the jaw comprise thinning of cortical bone, enlarged marrow spaces, generalized rarefaction of the alveolar bones, and coarse trabecula. In the parietal bones, the enlarged dipole and the thin cortex covering the coarse vertical trabecula result in a “hair-on-end” appearance. The extra medullary hematopoiesis result in pressure on the nerves, causing cranial nerve palsies [20].
No correlation between the skeletal, dental and chronological age was observed in β-thalassemia major patients [26]. The skeletal retardation increases with age due to endocrine hypofunction (secondary to iron deposition), hypoxia (from severe anemia) or toxic actions of iron enzyme systems, leading to tissue injury [27]. The enamel and dentin are the best indicators of iron deposition. Moreover, the deciduous and permanent teeth of thalassemia patients contain up to five times more iron concentration compared to that in normal individuals [20]. High iron concentration explains teeth discoloration in patients with β-thalassemia major [20].
Thalassemic patients do experience several secondary effects, which can affect their dental health in a number of ways. Some of the other oral considerations are prevalence of dental decay problems, pale-colored gums, burning sensation in tongue, painful swelling of salivary glands and dry mouth, reduced IgA resulting in reduced salivary protection, and etc.
Due to the clinical variability in signs and symptoms presented by the thalassemia patients, the main aspect of dental care is the necessity to provide it through a coordinated team approach, ensuring a close cooperation with the hematologist. In order to carry out complete risk assessment, detailed information on the patient’s recent blood test results and clinical status should be retrieved from the hematology team for guaranteed reduction in the risks involved in the planned dental care.
Besides the orofacial appearances associated with chronic anemia, patients may appear to be exhausted, lazy and poorly interested in physical activities [28]. Dental care should be modified according to their tolerance level on the day of treatment.
Thalassemic patients also experience frequent tooth decay problems due to the median salivary concentrations of IgA and phosphorous. In cases of intense dental caries, the risk of infections in the roots (or abscess) increases. At times, the infections spread to the tissues of face and neck that leads to extraction of the affected teeth resulting in loose gums and teeth structure [29].
The increased chance of infection should be considered during the dental treatment. Thalassemia patients undergoing splenectomy are at a massive risk of infection followed by bacteremia. Antibiotic prophylaxis must be given before performing invasive dental procedures [30].
Furthermore, these patients are also at a high risk of having AIDS or viral hepatitis due to repeated blood transfusion, and therefore, screening tests should be carried out at regular intervals. Teeth extraction should be carried out at the time of admission for blood transfusion, when the hemoglobin level is at its highest. Moreover, regular fluoride and prophylaxis applications are also recommended [30].
Iron overload, also known as hemochromatosis, is one of the common complications of thalassaemia. This hereditary disorder of metabolism can lead to organ damage and mortality [31,32], if left undiagnosed and untreated. Iron deposition in the parenchymal tissues of thalassemia patients can be observed within a year of the onset of regular blood transfusions [33]. Studies have shown that iron gets accumulated in the cardiac, endocrine, hepatic tissues and gingivae of thalassemia major patients [34,35]. While the impact of iron deposits on periodontal health is unidentified, more studies investigating the use of gingival biopsies for the diagnosis of iron overload are required. It is, thus, advised that dentists should be extra-attentive in case of dental problems in thalassemia patients. The oral ailments may worsen with increase in the iron load [29].
Individuals with thalassemia are at a high risk of carriage of HIV and Hepatitis B, C, and G viruses. Invasive dental care in thalassemia patients receiving regular exchange transfusion should be scheduled within a week of the planned transfusion, as the patient’s blood counts would be optimal [28].
Chronic anemia can result in cardiomyopathy and get further exacerbated by cardiac iron overload [36]. Although patients may be asymptomatic with their cardiac dysfunction when experiencing a stressful dental procedure and/or being anxious, they may rapid their cardiac symptoms [28]. Studies have also deduced that the bacteria causing periodontal problems can move into the bloodstream and result in vasculitis, thereby, contributing to heart disease and stroke [37].
During the dental procedures of patients with cardiac problems, the dentists should be decisive of using anesthesia. Epinephrine content in anesthesia may result in cardiovascular problems in patients with heart ailments, which involves high blood pressure, angina (chest pain), heart attack and arrhythmias [38]. An orthodontist needs to be aware of the degree of cardiac involvement and should use appropriate precautions accordingly.
Utmost attention should be focused on the obtainment of as much information as possible on thalassemic patients for proper distinction before subjecting to a guaranteed safe dental management. Therefore, the type of thalassemia must be recognized, accompanied by the treatments received, the degree of iron accumulation-related organ involvement, and consequently, the patient’s prognosis and life expectancy.
The medical diagnosis is of great aid in the planning of dental treatment as it varies according to the type of thalassemia involved. Thalassemia trait patients can be considered for more complex treatment modules. A simple case history documented by the dental expert is, therefore, important for dental treatment, which can be suitable as per the patient’s oral health and quality of life.
As in any patient with chronic anemia, poor healing may ensue after surgical dental procedures. The possibility of exacerbated symptoms of cerebral or cardiac hypoxia persists in an anemic patient in the case of substantial bleeding; hence, surgery is the best possible option for successful treatment of the facial deformities.
All the dental procedures of thalassemic patients should be conducted under the guidance of a hematologist and should not collide with the patient’s blood transfusion schedules. The patients with low hemoglobin level are mostly not recommended with oral surgeries. The side-effects of the prescribed medications to the thalassemia patients should be considered before-hand [39].
Hematological disorders frequently affect both the soft and hard tissues of mouth with diverse characteristics. Oral manifestations include color modifications of the mucosa and hypertrophy of gingiva, mucosal destruction in the form of ulceration, bleeding and hemorrhage, tooth color, lymph node, decreased bone density, and enlarged marrow space. Advances in the treatment options resulted in long-term disease-free survival, and appropriate dental and periodontal maintenance improved the patient’s quality of life. Preventive dental care is a prime necessity for the thalassemia patients, and the treatment procedure should be modified according to the patient’s need to minimize any physical damage and for better treatment outcomes.
Dentists should have in-depth understanding about the proper management of dental problems of thalassemic patients. Multidisciplinary approaches should be implemented by dental experts in association with hematologists for safe dental treatment of these patients.
The authors are thankful to www.manuscriptedit.com for providing English language editing and proofreading services for this manuscript.

![]()

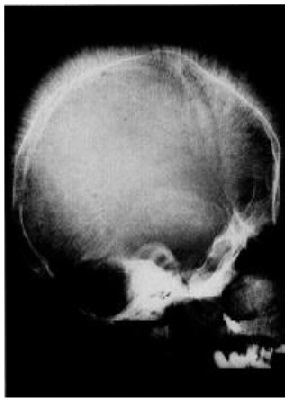 |
Figure 1: Radiographs for Cooley facies |
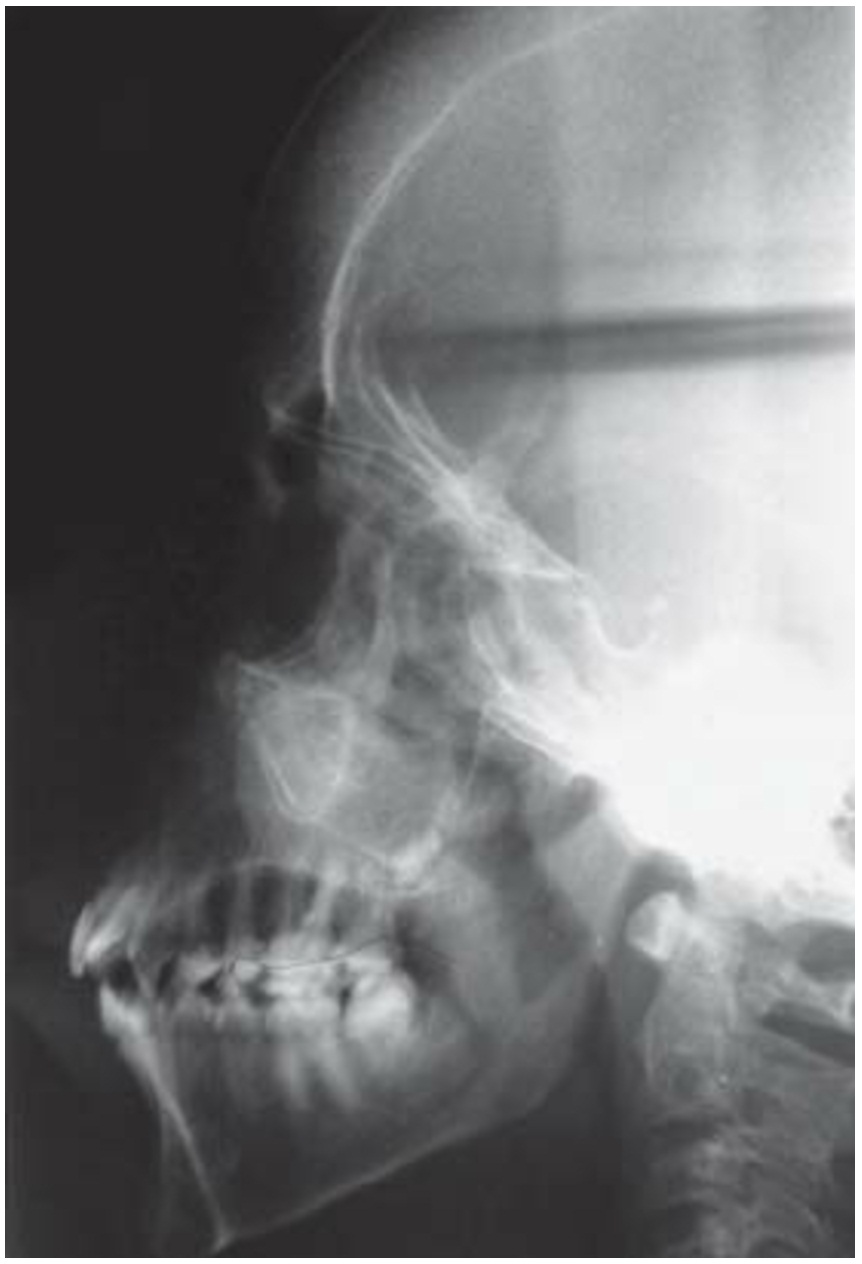 |
Figure 2: Maxillary teeth spacing and malocclusion |
Silent Carrier |
Alpha (α) or beta (β) thalassemia trait |
Severe β-thalassemia (Colley’s anemia) Thalassemia major (transfusion dependent) Thalassemia intermedia (no regular transfusions required) |
Hemoglobin H disease (HbH disease) |
Fetal hydropexis |
Table 1: Clinical classifications of thalassemia [9] |












