Open Access
Research Article
Max Screen >>
ISSN: 2455-7641
Copyright: © 2015 Michiels JJ. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
The WHO defined JAK2V617F mutated myeloproliferative neoplasms (MPN) consist of normocellular essential thrombocythemia (ET), ET with features of early PV (prodromal PV), ET with hypercelular megakaryocytic granulocytic myeloproliferation (ET.MGM), and various stages of polycythemia vera (PV) when the WHO and European Clinical and Pathological (WHO-ECMP) criteria are applied. Bone marrow cellularities on histopathology are overlapping in normocellular ET, prodromal PV and PV carrying the JAK2V617F mutation. Increased erythrocyte counts at a cut off level of 5.8x1012/L separates JAK2 mutated ET and prodromal PV from overt PV. JAK2 wild type ET and MF carrying the MPL515 mutation is a distinct MPN entity featured by increase of clustered small and giant megakaryocytes with hyperlobulated stag-horn-like nuclei, in a normal cellular bone marrow without features of PV. JAK2/MPL wild type ET and MF carrying one of the calreticulin (CALR) mutations is the third distinct entity associated with prefibrotic primary megakaryocytic and granulocytic myeloproliferation (PMGM) characterized by dense clustered, large, and immature dysmorphic megakaryocytes with bulky (bulbous) hyperchromatic nuclei, which are never seen in JAK2V617F mutated ET, prodromal PV, EMGM and PV, and also not in MPL515 mutated ET and MF.
Keywords: Essential thrombocythemia; Polycythemia vera; Myelofibrosis; Bone marrow pathology; JAK2 mutation; MPL mutation; Calreticulin mutation
As the PVSG classification provided no strict definitions for early stage essential thrombocythemia (ET) and polycythemia vera (PV), we defined in 1980 the Rotterdam Clinical and Pathological Criteria (RCP) for the diagnosis of ET and PV (Table 1) [1]. Michiels & Thiele improved the RCP criteria for ET and PV and defined the European Clinical and Pathologic (ECP https://www.mpn-stichting.nl/doctors_brochure_2004.pdf) for ET, PV and hypercellular ET with features of prefibrotic myelofibrosis (MF) [2-8]. Thiele joined the WHO to define the 2008 WHO classification of the myeloproliferative neoplasms (MPN) ET, PV and primary myelofibrosis (PMF) [9,10]. In response to the significant shortcomings of the 2008 WHO classification, we could much better define the WHO and European Clinical Molecular and Pathological (WHO-ECMP) [11-13] criteria by the combined use of bone marrow histology, biological laboratory markers and JAK2V617F mutation screening to distinguish the broad spectrum of JAK2V617F mutated trilinear MPN entity (Tables 2 and 3) to clearly separate JAK2 mutated MPN from JAK2 wild type MPN carrying one of the MPL or calreticulin (CALR) mutations (Tables 4 and 5) [14,15]. The 2008 WHO defined prefibrotic JAK2V617F mutated myeloproliferative neoplasms (MPN) can be subclassified and staged as normocellular essential thrombocythemia (ET), ET with features of early PV (prodromal PV), ET with hypercelular megakaryocytic granulocytic myeloproliferation (ET.MGM), and various stages of polycythemia vera (PV) when the WHO-ECMP criteria are applied (Tables 2 and 3). In this report we could distinguish JAK2V617F positive ET and prodromal PV from JAK2/wild type normocellular ET carrying the MPL515 as a distinct MPN entity with no features of PV. JAK2/MPL wild type CALR mutated hypercellular ET is associated with prefibrotic primary megakaryocytic granulocytic myeloproliferation (PMGM) and appears to be the third MPN entity of ET and MF without features of PV.
Hemoglobin, hematocrit, erythrocytes, leukocytes, platelets, iron status and chemical parameters were routinely performed. Erythrocyte volume (red cell mass RCM) was measured using Cr51 (natriumchromate) labeled autologous erythrocyte and plasma volume was measures with J131-human serum albumin. The leukocyte-alkaline-phosphatase (LAP) stain was performed with Na-naphtyl and Fast Garnet G.B.C Salt. Iron stain of bone marrow smears were performed with Prussian blue reagens. Bone marrow biopsies from the iliac crest were stained with hematoxylin and eosin for histopathology evaluation. All bone marrow biopsies were evaluated by expert hematopathologists for morphology, grading of cellularity and scoring of reticulin fibers according to PVSG recommendations [16] (Table 1). Our pathologists used the collagen staining of Mason for objective detection of collagenisation of reticulin fibers to clearly distinguish between early stage reticulin fibrosis (RF) from advanced reticulin/collagen fibrosis for grading of myelofibrosis (MF, Table 1C).
In the period of January 1975 to December 31, 1980, we prospectively studied 30 consecutive early prefibrotic stage MPD patients, who presented with erythromelalgic thrombotic thrombocythemia (ETT), 14 ET and 16 PV patients (Table 6). The mean age of 30 ETT patients (ET and PV) was 56.7 (range 33-96) years. Eleven of 14 ET patients had platelet counts below 1000x109/L. Spleen size on scan was slightly increased in 5 of 14 ET and in 13 of 16 PV patients. Leukocyte count counts was increased (>10x109/L) in 5 out of 14 patients with ET and in 14 of 16 PV patients. LAP score was increased (>100) in 12 out of 14 ET and in all PV patients.
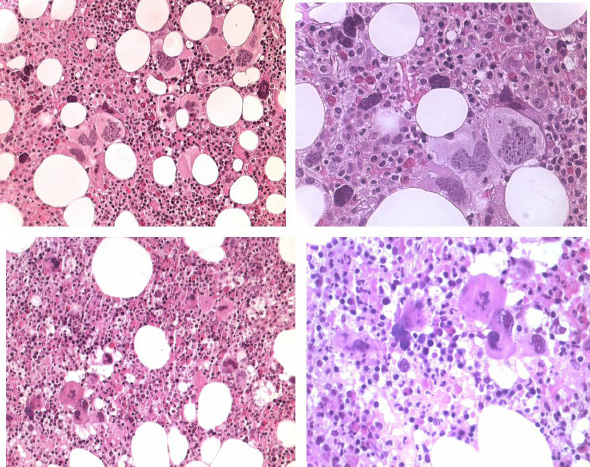
Increase of clustered large pleomorphmegakaryocytes in bone marrow smears and biopsies was diagnostic for MPD in 14 ET and 16 PV patients (Table 6) (Figures 1 and 2). A normocellular bone marrow picture (cellularity <60%) with increase of clustered pleomorphic megakaryocytes and no increase of cellularity (Figure 1) was seen in 7 of 14 ET and in one of 16 PV patients.
A moderate increase of cellularity (1+ = 60-80%) in the bone marrow due to increased erythropoiesis leading to a ET/PV picture was seen in 3 ET and 4 PV patients (Figure 2). A typical PV bone marrow picture with pronounced increase of cellularity (2+ = 80-100%) due to predominantly increased erythropoiesis or trilinear hypercellular hematopoiesis due to erythro/megakaryo/granulocytic myeloproliferation (EMGM) was seen in 2 of 14 ET patients and in 11 of 16 PV (Figure 2). These results indicate that bone marrow histopathology on its own is not reliable to differentiate between ET and PV but appeared to be a powerful tool to differentiate ET and PV from all variants of primary or secondary erythrocytosis and reactive thrombocytosis with a sensitivity and specificity near to 100%. The morphology of large pleomorphic megakarocyteswere not different in ET and PV. Pleomorphism of megakaryocytes became more pronounced in the hypercellular (80-100%) bone marrows of advanced PV (Figure 2).
RCP defined ET and PV patients show overlapping bone marrow pictures with increase of pleomorphic clustered megakaryocytes and varying degrees of increased erythropoisesis at time of diagnosis (Figures 1 and 2). The correlation curves between erythrocyte count, hemoglobin (Hb) or hematocrit (Ht) versus red cell mass (RCM) showed the best correlation between erythrocyte counts and RCM (Figure 3). Increased erythrocyte counts above 5.8x1012/L is diagnostic for PV (Table 1B) in the presence of large pleomorphic megakaryocytes in a bone marrow smears and biopsy. Increase of erythrocytes appears to be independent from the iron deficient status and persists in PV in a clinical remission obtained by repeated venesection (Figures 4 and 5). Two ET patients (A1 and A11, Table 6) had borderline increased RCM, which was associated with erythrocytes above 5.8x1012/L, increased LAP score, and a negative bone marrow iron stain. In retrospect both cases should be reclassified as PV patients. The ET case (A6) with an erythrocyte count of 6.0x1012/L and increased LAP score developed PV within 2 years. Bone marrow iron stain was negative in all PV patients (N=16) and in 4 out of 12 evaluable ET patients.
Between 1997 and 2007 we prospectively studied 10 JAK2V617F mutated patients, who presented with erythromelalgia or migraine-like microvascular cerebral ischemic attacks (MIA) and were referred for expert evaluation and treatment recommendation (Table 7). The clinical diagnoses were ET in 6 and PV in 4 without the use of bone marrow histopathology (Table 7A). The 6 ET were heterozygous for the JAK2V617F mutation. Three PV patients were homozygous for the JAK2V617F mutation (Case 7, 9 and 10, Table 10 in Table 7). All ET patients had an erythrocyte count below 5.8x1012/L and all PV had erythrocyte counts above 6x1012/L (Figures 4, 5 and 6) (Table 7A).
The clinical diagnosis of the 10 JAK2V617F positive patients without the use of bone marrow histology data was ET in 6 and PV in 4 cases (Figures 4, 5 and 6) (Table 7A).
The diagnosis of the 10 JAK2V617F positive MPN patients based on bone marrow histology picture alone as blindly judged by pathologists and was consistent with ET in 3 and PV in 7 cases (Table 7B). The 3 ET patients diagnosed as PV bone marrow histology evaluation had very low serum EPO levels and EEC, but erythrocyte counts less than 5.8x1012/L consistent with the diagnosis of prodromal PV (Table 7B).
The diagnoses according to 2008 WHO criteria were ET in 5, MPN unclassifiable in 1 and PV in 4. The diagnoses according to the WHO-ECMP criteria for diagnosis and staging of ET and PV patients (Tables 2 and 3) were normocellular ET in 2 cases, prodromal PV (ET with low serum EPO, the presence of EEC and normal erythrocyte counts) in 3 cases, ET with a MGM bone marrow histopathology (ET.MGM Table 7B) with no leuko-erythroblastosis in 1 case, and acute onset PV in 4 patients (Cases 7, 8, 9 and 10, Table 7B). Examples of bone marrow histopathology of normocellular ET and prodromal PV are shown in figure 4 and of PV in figure 6 demonstrating that bone marrow histopathology alone cannot distinguish JAK2V617F mutated ET, prodromal PV and PV.
Bone marrow cellularity of the 10 JAK2V617F mutated MPN patients (6 ET and 4 PV) ranged from range 60% to 90% (Table 7B).There was an increased erythropoiesis in 8/10 and increased granulopoiesis in 4/10 patients. Interestingly, 3 ET with early features of PV (prodromal PV) fulfilled the bone marrow features of prodromal PV and 4 cases presented with rapid onset PV (Cases 7, 8, 9 and 10). The three ET patients with prodromal PV developed overt PV after long-term follow-up of 8, 9 and more than 10 years (slow onset PV, Table 7). Myelofibrosis (MF) was scored according to Table 1 Case MF-0 (RF-0/1) in 8 (5 ET, 3 PV), and MF-1 (RF-2) in 2 (1 ET, 1 PV) MPN patients. ET case 5 showed increased megakaryocytic-granulocytic myeloproliferation (MGM) bone marrow histology consistent with the ET.MGM-entity (Table 7B).
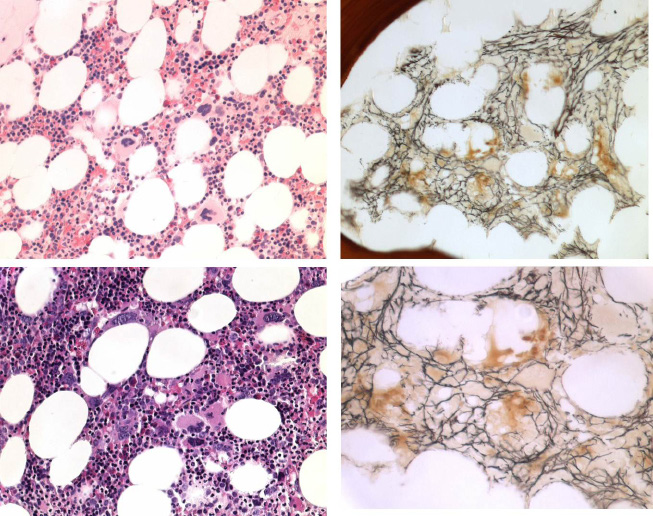
JAK2 wild type ET carrying the MPL515 mutation: In 2008 we studied bone marrow histopathology in 12 cases with JAK2 wild type ET carrying the MPL515 mutation kindly provided by the courtesy of Dr. Vannucchi, Florence, Italy. Bone marrow histology from a recent patient with JAK2 wild type ET carrying the MPL515 mutation consistently displayed clusters small and large megakaryocytes with a greater number of giant megakaryocytes with hyperlobulated stag-horn nuclei in a normal cellular bone marrow and no increase of erythropoiesis as compared to JAK2V617F ET with features of PV in the bone marrow (Figure 7). Bone marrow histopathology in MPL515 mutated ET (Figures 8, 9 and 10) versus WHO-ECMP defined JAK2V617F mutated ET were different on 4 points.
First, The presence of clustered small and giant megakaryocytes with deeply lobulated stag-horn like nuclei (Figures 7, 8 and 9) in ET carrying the MPL515 mutation are not seen in JAK2V617F positive ET, prodromal PV, ET.MGM and PV. The pleomorphic megakaryocytes in JAK2V617F mutated ET in bone marrow smears and bone marrow biopsy were not larger but similar in size with small, medium and large megakaryocytes (pleomorphic) as in PV (Figure 4 and 6).
Second, there was local increase of erythropoiesis in areas of loose clustered pleiomorphicmegakaryoctyes in JAK2V617F mutated normocelluar ET and in prodromal PV (Figure 7).
Third, JAK2 wild type ET carrying the MPL515 mutation have no clinical, laboratory and bone marrow features of prodromal PV at diagnosis, do not evolve into overt PV during follow-up, and have normal serum EPO and ferritin levels.
Fourth, we observed significant increased reticulin fibrosis grade 2 to 3 in MPL515 mutated ET in bone marrow areas of normocellularity but with dense clustered megakaryocytes (Figure 10), which is not seen in JAK2V617F mutated normocellular ET, hypercellular prodromal and PV (Figure 10).
JAK2-MPL-wild type CALR mutated PMGM: From 1994 to 2006, we documented a case of JAK2 wild type ET with a PMGM bone marrow (Figure 11) in a 9-year-old boy (referred to us from Basel, Switserland) with a platelet count of 1596 to 1946x109/l, no splenomegaly on palpation, white blood differential count metamyelocytes 0.5%, banded forms 1%, segmented granulocytes 52%, basophiles 2.5%, lymphocytes 35% monocytes 6%, low LAP score, and a hypercellular (80-100%) bone marrow with a predominant prefibrotic primary megakaryocytic and granulocytic myeloproliferation (PMGM, Table 5), absence of reticulin fibers, loose to dense clustering of large dysmorphic megakaryocytes variable in size with cloud-like hypoploid nuclei. The dysmorphic megakaryocytes show definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear cytoplasmic ratio (Figure 11, arrows), which are not seen in JAK2 wild type ET carrying the MPL515 mutation and also not in prefibrotic JAK2V617F mutated ET, prodromal PV and PV. Such bone marrow findings of PMGM are consistent with pronounced ET as the presenting feature of prefibrotic PMGM, and do not meet the criteria for fibrotic PMF according to the 2008 WHO classification (Table 5) [15,18]. The 10 years follow-up from 1994 to 2004 showed normal blood cells counts, absence of the JAK2V617F mutation, no evidence of myelofibrosis, and no splenomegaly on palpation (Figure 11).
Bone marrow histology in CALR mutated JAK2 wild type ET and MF: We prospectively observed in 2004 a case of WHO-ECMP defined PMGM (Table 5) (Figure 12) in a 37-years old woman (asymptomatic except fatigue) with JAK2 wild type (anno 2006) hypercellular ET: platelets 1205 x109/L, Hemoglobin 12.5 g/dl, leukocytes 18 x109/L, borderline LDH, spleen size 13 cm on echogram (normal value <12cm) as the presenting features of primary megakaryocytic granulocytic myeloproiferation (PMGM, RF-1, Figure 12). This early fibrotic stage of JAK2 wild type hypercellular ET associated with PMGM developed anemia, significant splenomegaly and myelofibrosis during 10 years of follow-up and appeared to be CALR positive when tested in 2014.
In 2014/2015 we prospectively studied seven consecutive newly diagnosed CALR positive hypercellular ET or MF and found consistent bone marrow characteristics of hypercellular thrombocythemia as the presenting feature of prefibrotic and early fibrotic stages of JAK2 wild type PMGM. Four of these seven CALR mutated MPN patients presented with aspirin sensitive microvascular disturbances of erythromelalgic, cerebral and ocular ischemic manifestations (Sticky Platelet Syndrome) as the specific presenting manifestations of myeloproliferative thrombocythemia. Bone marrow histology in prefibrotic cases of CALR ET (Figure 13 left), and in early fibrotic stage CALR MF (Figure 13 right) show dysmorphic megakaryocytes with definite abnormalities of maturation with bulky (bulbous) hyperchromatic nuclei and some disturbances of the nuclear cytoplasmic ratio consistent with CALR mutated PMGM, which are not seen in MPL515 mutated ET and also not in JAK2V617F mutated ET, prodromal PV and classical PV.
Recently, we observed a case of aleukemic splenomegaly in a 67 year-old-man, who presented in January 2015 with anemia with normocytic anemia (hemoglobin 5.2 mol/l, hematocrit 0.25 and erythrocytes 3.1x1012/L, platelets 265x109/L, leukocytes 6.5x109/L) weight loss from 84 to 71 kg in the last 2 years, minor fatigue, no sweatings and asymptomatic splenomegaly on physical examination (Figure 14). The spleen length on echogram was 24 cm. Morphology of periperal blood smear showed anisoctytosis, trear drop erythrocytes and spherocytes, white blood cell differential counts was normal with the presence of banded (2-4%), blasts (1-2%), myelocytes (2-5%) and erythroblasts (3-4% of WBC). There was a drytap on bone marrow aspiration. Bone marrow biopsy revealed a strongly fibrosed hematopietic stroma with numerous dysmorphic dense clustered dysmorphic megakaryocytes, with remnants of previous erythropietic and myeloid precursor cells and reticulin fibrosis grade 3 to 4 (Figure 14), which could be diagnosed as JAK2 wild type calreticulin (CALR) mutated MF without a history of ET. This CALR mutated MF must have had a long history of subclinical CALR MPN as Michiels & Ten Kate already have demonstated in the 1980s (Table 8) [17]. In 1992, we analysed the long-term follow-up of a 61-year-old man, who presented in 1971 with primary myeloid metaplasia of the spleen initially diagnosed as prefibrotic primary myeloproliferative disease (MPD) upon bone marrow histology (Table 8) [17]. The spleen size increased from 18 cm to 23 cm length diameter on scan (normal value <12cm) in 1978 and 1988, but the peripheral blood counts remained normal for 14 years of follow-up (Table 8). From 1985 to 1989 he suffered from aspirin responsive erythromelalgia of fingers and toes at platelet counts of 421x109/L. After 17 years of follow-up this case of PMD developed in 1988 anemia, thrombocythemia, and symptoms related to splenomegaly consistent with the diagnosis of agnogenic myeloid metaplasia and advanced myelofibrosis (MF 2/3).
In 1940, Dameshek believed that the following minimal data should be present before a definite diagnosis of PV can be made: plethoric appearance, splenomegaly, definitely elevated erythrocyte count above 6x1012/L, elevated platelet count, and elevated hematocrit (Table 1) [18]. The bone marrow is diagnostic showing increased trilinear hematopoiesis and large megakaryocytes. ET and PV patients present identical (typical) ET/PV or PV bone marrow histology pictures and the most objective criterion to differentiate ET from PV appeared to be the erythrocyte count at a cutoff level of 5.8x10/12L obviating the need to measure red cell mass (RCM) (Tables 2 and 3) (Figures 3 and 5). We did not observe a significant difference in the morphology of pleomorphic megakaryocytes within the prefibrotic JAK2V617F mutated MPNs normocellular ET, prodromal PV and PV. In JAK2V617F positive EMGM (or masked PV), the pleomorphic megakaryocytes show dysmorphic features of nuclei (not cloud-like), which is clearly in between ET and post-ET myelofibrosis (Table 2). There is significant overlap of serum EPO levels in PV versus control and controls versus increased serum EPO in congenital erythrocytoses [11-13]. A low serum EPO level has been described in about half of PVSG defined ET patients, who are predicted to carry the JAK2V617F mutation and can be regarded as prodromal (forme frusta) PV [11,12]. The diagnostic impact of low serum EPO (ELISA assay) below the normal range (<3.3IU/L) had a sensitivity, specificity and positive predictive value of 86%, 97% respectively for the diagnosis of PV [13]. Piche, et al described the bone marrow histopathology findings in 59 JAK2V617F positive ET and 44 JAK2 wild ET cases [16], which are completely in line with the present study that ET patients with JAK2V617F mutation indeed have PV-like morphological bone marrow changes of pleomorphic large megakaryocytes. Our findings in WHO-ECMP defined ET, prodromal PV patients and PV patients with increased erythrocytic, megakaryocytic and granulocytic myeloroliferation (JAK2V617F-EMGM) as well as increased serum LDH levels and spleen size were seen in more pronounced cases JAK2V617F positive ET in particular at higher JAK2V617F mutation load [16].
Our bone marrow findings of JAK2 wild type ET patients carrying the MPL515 mutation displayed small and giant megakaryocytes with a greater number of large deeply lobulated stag-horn nuclei and more dense clustered megakaryocytes in a normocellular bone marrow with no increase of erythropoiesis. JAK2 wild type MPL515 mutated ET has no clinical, laboratory and bone marrow features of prodromal PV at diagnosis and does not evolve into overt PV during follow-up [19-21]. JAK2 wild types ET carrying the MPL515 mutation have normal serum EPO and ferritin levels and no spontaneous endogenous erythroid colonies (EEC). The prevalence of ET or MF patients carrying the MPL515 mutation may range from 5 to 10% of the JAK2 wild type MPN population. In a large collaborative European study, Jones, et al. [21] investigated a cohort of 176 MPN cases with the MPL515 mutation: W515L in 110 and W515K in 58. The overall mutation levels were lower (25%) in W515L (N=106) than the level of 37% in cases with W515K (N = 32). Of the 138 cases (ET, N = 99; MF, N = 36), the median W515L mutation levels were significantly lower (21%) in ET than those (46%) in MF. The 29 homozygous MPL515 positive cases had a diagnosis of MF in 15 and ET in 12 patients.
JAK2 wild type prefibrotic primary megakaryocytic granulocytic myeloproliferation (PMGM) according to the 1990 Hannover Bone Marrow Classification [22-24] has been labeled as prefibrotic primary myelofibrosis (PMF) in the 2008 WHO classification [10] and represent the third distinct MPN entitiy [22]. In this report, JAK2/MPL wild type, hypercellular ET is associated with a PMGM bone marrow (Table 5) (Figures 11, 12 and 13). The bone marrow findings in CALR mutated ET and MF in Figures 13 and 14 are consistent with WHO defined prefibrotic PMF and with WHO-ECMP defined PMGM (Table 5). The bone marrow features in CALR-MGM in two cases with hypercellular ET in Figure 13 clearly differs from the pleomorphic megakaryocytes in JAK2V617F mutated ET and PV and from giant megakaryocytes and hyperlobulated staghorn-like nuclei in MPL515 mutated ET [25]. The updated 2015 WHO-ECMP criteria in Tables 2, 3, 4 and 5 have the potential to properly diagnose and stage the each of the JAK2V617F, JAK exon 12, MPL515 and CALR mutated MPNs based on clinical features (splenomegaly, LDH, degree of anemia), relevant bone marrow histology criteria, grading of bone marrow fibrosis, constitutional symptoms and laboratory features including EEC, EPO levels, and mutation load (Figure 15) [15,25]. The broad spectrum and its various clinical evolution patterns in terms of primary hematopoietic neoplasms of the spleen and secondary fibrosis of the bone marrow in JAK2V617F, MPL515- and CALR-mutated MPN has to be further delineated in prospective clinical and basic research studies.
We gratefully thank Dr. Juergen Thiele for his significant contribution to the formulation of the ECP, WHO and ECMP criteria for the MPNs ET, PV, PMF and PMGM and its differentiation from Ph+ ET, Ph+ CML, and the Ph-negative CMML, MDS and CNL just by bone marrow histopathology alone [6-8,10]. We thank the molecular biologists Dr. M. Bakkus, University Hospital Brussels and Dr. P.J.M. Valk, Erasmus University Hospital Rotterdam for screening of MPN patients for the JAK2V617F and CALR mutations.

![]()

 |
| Figure 1: Presence of large platelets in peripheral (EDTA) blood smear (left top), increase of clustered enlarged megakaryocytes in a normocelluar ET bone marrow with stainable iron (left bottum). Local increase of erythropiesis (bold arrows) in areas of loose clustered pleomorphic megakaryocytes in patients with essential thrombocythemia: ET bone marrow picture [1] |
 |
| Figure 2: ET (left upper), ET/PV (left right), and PV (left bottum) bone marrow features in ET and PV patients. Pleiomorphic megakaryocytes in ET (upper panels) have less hyperlobulated nuclei as compared to PV (left bottum). Dense clustered pleiomorphic megakaryocytes in PV/RF (right bottum) in advanced PV show dysmorphic nuclei [1] |
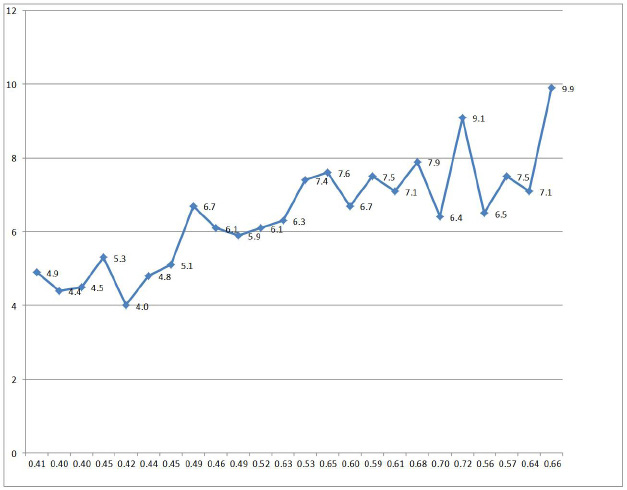 |
| Figure 3: Correlation of erythrocyte counts and erythrocyte volume or red cell mass in 5 ET and 13 PV patients. Two ET patients with borderline increased RCM had erythrocyte counts above 5.8x1012/L associated with a typical PV bone marrow histology and should in fact be diagnosed as PV. Erythrocyte count at a cut off level of 5.8x1012/L differentiates between ET and PV even in iron deficient state of PV remission after phlebotomy |
 |
| Figure 4: Case 2 in Table 7 with a 10 to 5 years history of stable JAK2V617F positive ET disease and ET bone marrow picture in 2006: normocellular ET stage 1 showing a typical ET bone marrow picture with cellularity of 60% due to slight increase of erythropiesis (upper panels). Case 6 in Table 7 with ET and a PV bone marrow in 1997, who developed PV seven years later in 2004 with a good response to interferon in 2005 (lower panels) |
 |
| Figure 5: Case 7 in Table 7 presented with rapid onset JAK2V617F positive erythrocythemic PV with a typical PV bone marrow picture in 2005 carrying homozygous JAK2V617F mutation on repeated testing. The erythrocyte counts were far above 6x1012/L and remained above 6x1012/L at time of hematological remission of PV for several years by phlebotomy alone on top of low dose aspirin |
 |
| Figure 6: Rapid onset trilinear JAK2V617F homozygous positive PV Case 9 in Table 7 with a typical trilinear PV bone marrow picture complicated by bleeding, poor response to pegylated interferon indicating the need to treat with hydroxyurea on top of low dose aspirin resulting in eliminating the thrombo-hemorrhagic diathesis and significant imporovement of quality of life during long-term follow-up (2006-2013) |
 |
| Figure 7: 65 years old man with RBC 5.37x1012L, HB 15.8 g/dL, MCV 89, WBC 12.0 x10/9/L, Plts 517x109L, LDH 600 UI/L and JAK2V617F mutation (allele burden: 20% on peripheral blood). Diagnosis PV according WHO-ECMP (left). 55 years old woman with MPL515-positive normocellular ET with normal values for hemoglobin, hematocrit, leukocytes and platelet around 1000x109/L. Dr. Campr: small medium, large and giant megakaryocytes with staghorn-like hyperlobulated nuclei (right) |
 |
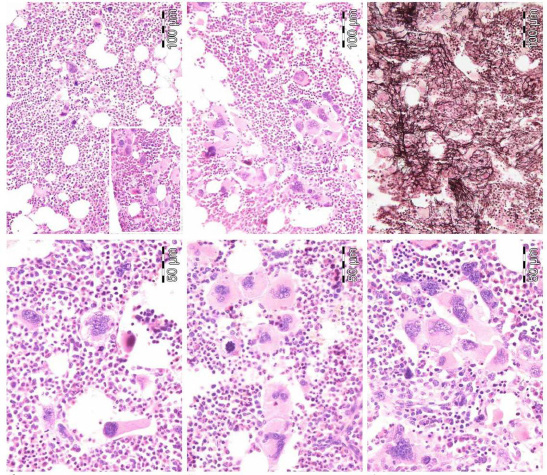
| Figure 8: JAK2 wild type ET carrying the MPL515 mutation with enlarged and giant mature megakaryocytes with loose clusters of hyperlobulated, "stag-horn" hyperlobulated nuclei. Case 1 and 2, Dr. Vannucchi, upper panels. Case 3, Dr. Vannucchi lower panels |
 |
| Figure 9: JAK2 wild type, MPL515 mutated ET with enlarged and giant mature megakaryocytes with loose clusters of hyperlobulated, "stag-horn" hyperlobulated nuclei. Case 4, Dr. Vannucchi, upper panels. Case 5, Dr. Vannucchi, lower panels |
 |
| Figure 10: Case of JAK2 wild type MPL515 mutated ET with clustered large and giant megakaryocytes and increase of reticulin fibrosis grade 2 (RF 2). Such increase of RF 2 in a normocellular bone marrow is not seen in JAK2V617F mutated ET and PV, and also not in JAK2 wild type PMGM(Figures 11 and 12) and CALR-MGM (Figures 13 and 14) |
 |
| Figure 11: Third MPN entity presenting with JAK2 wild type ET associated with prefibrotic primary megakaryocytic and granulocytic myeloproliferation (PMGM, colour picture), which is characterized by a hypercellular bone marrow due to dual myeloproliferation of granulopoiesis and dense clustered enlarged immature dysmorphic megakaryocytes with bulky (bulbous) hyperchromatic nuclei (arrows), which are never seen in JAK2 wild type MPL515 mutated ET and also not in the prefibrotic JAK2V617F mutated ET. During long-term follow-up, reduction of platelet count to normal or near normal by treatment with hydroxyurea in 1994 followed by anagrelide from 1995 to 1998 the bleeding manifestations did not recur. After discontinuation of anagrelide in 1998 the patient remained asymptomatic, the platelet counts were between 600 and 800x109/L, which normalized after 8 years of follow-up. From 2001 to 2005 haemoglobin and hematocrit reached completely normal values |
 |
| Figure 12: 37-years old woman (asymptomatic except fatigue) with JAK2 wild type hypercellular ET (anno 2006): platelets 1205 x109/L, Hb 12.5 g/dl, leukocytes 18 x109/L, borderline LDH, spleen size 13 cm on echogram (normal value <12cm) as the presenting features of primary megakaryocytic granulocytic myeloproiferation (PMGM, RF-1) diagnosed according ECMP criteria but not meeting the 2008 WHO criteria for primary myelofibrosis (PMF). In 2015 this case proved to be calcireticulin (CALR) mutated |
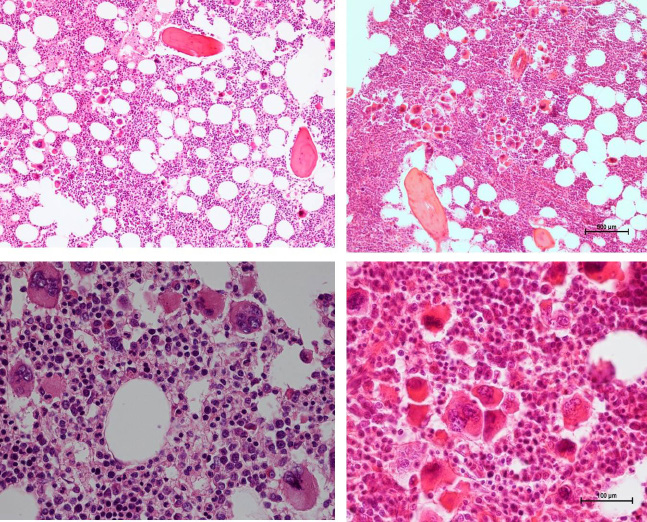 |
| Figure 13: Bone marrow features in newly diagnosed calreticulin (CALR) mutated essential thrombocythemia (ET, left) and early stage myelofibrosis (MF, right) Left: Clinical case of JAK2/MPL negative and calcireticulin (CALR) positive ET who presented with normal vlaues for hemogobin, hematocrit and erythrocytes, platelet count of 1832x109/L and slight splenomegaly (16 cm lenght diameter on echogram). Bone marrow histology is hypercellular with relative decrease of erythropoisis, dense cluster of immature megakaryocytes with hypolobulated nuclei consistent typical PMGM and no increase of reticulin fibrosis (Table 5) Right: Clinical case of CALR positive myelofibrosis (MF): hemoglobin 11.2 g/dL,hematocrit 0.33, leukocytes 9.2x109/L, platelets 347x109/L, LDH 1393 U/l, and the presence of tear drop erythrocytes, poikolocytosis and polychromasie in a peripheral blood smear, and hypercellular bone marrow with relative decrease of erythropoisis, dense cluster of immature megakaryocytes with hypolobulated nuclei consistent with PMGM, and reticulin fibrosis (RF) grade 2.The marrow histology features similar to WHO-defined primary myelofibrosis (PMF) and WHO-ECMP-defined PMGM or CALR-MGM, but distinct from JAK2V617F mutated ET and PV, and distinct from MPL515 mutated ET |
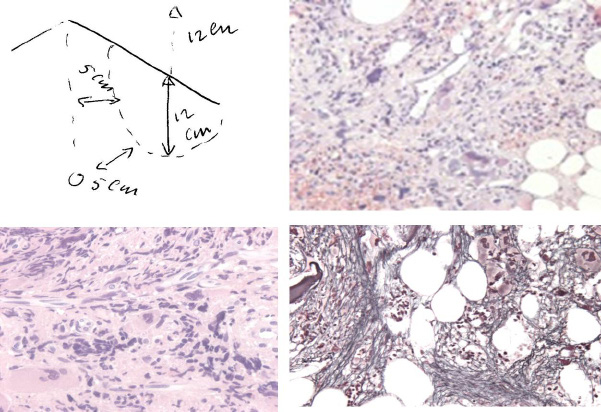 |
| Figure 14: A 67 year old man presented in 2015 with normocytic anemia (hemoglobin 5.2 mol/l, hematocrit 0.25 and erythrocytes 3.1x1012/L, platelets 265x109/L, leukocytes 6.5x109/L) weight loss from 84 to 71 kg in the last 2 years, minor fatigue, no sweatings, asymptomatic splenomegaly and dense clustered dysmorphic megakaryocytes, reticulin fibrosis grade 3 to 4, which was diagnosed as JAK2 wild type calcireticulin (CALR) mutated MF without a history of ET |
 |
| Figure 15: 2015 WHO-ECMP Myeloproliferative Neoplasms Classification and Transitional states |
| A. The 1980 RCP major (A) and confirmative (B) criteria for prefibrotic ET [1] | |
|---|---|
| A1 | Persistent platelet count in excess of 400x109/L. |
| A2 | Increase and clustering of enlarged megakaryocytes in bone marrow biopsy. |
| A3 | No or slight increase of reticulin fibers (RF 0 or RF 1) |
| B1 | Presence of large platelets in a peripheral blood smear |
| B2 | Absence of any underlying disease for reactive thrombocytosis and normal ESR. |
| B3 | No or slight splenomegaly on palpation or scan (<15 cm) |
| B4 | Increase of LAP-score and no signs of fever or inflammation |
| Exclusion criterion Ph+ chromosome and any other cytogenetic abnormality in blood or bone marrow cells |
|
| B. The 1980 RCP major (A) and minor (B) criteria for prefibrotic PV [1] | |
|---|---|
| A1 | Raised red cell mass. Male >36 ml/kg, female >32 ml/kg consistent with erythrocyte count of >6x1012/L (Dameshek 1940 [18]) |
| A2 | Absence of primary or secondary erythrocytosis by clinical and laboratory tests. |
| A3 | Slight, moderate or marked increase in bone marrow biopsy of clustered, enlarged pleomorphic megakaryocytes with hyperlobulated nuclei and moderate to marked increase cellularity of megakaryopoiesis/erythropoiesis or typically trilinear mega-erythro-granulopoiesis. |
| B1 | Thrombocythemia, persistant increase of platelet >400x109/L |
| B2 | Leukocytosis, leucocyte count >109/L and low erythrocyte sedimentation rate (ESR) |
| B3 | Raised leukocyte alkaline phosphatase (LAP) score >100, absence of fever or infection |
| B4 | Splenomegaly on palpation or on isotope/ultrasound scanning |
| A1+ A3 plus one of B establishes PV and excludes any variant of erythrocytosis | |
| C. Grading of bone marrow reticulin fibrosis (RF) and grading of myelofibrosis (MF)[5-8] | ||
|---|---|---|
| Grading reticulin fibrosis (RF) |
Grading MF Thiele, et al. | Description of reticulin fibers (RF) and reticulin/collagen fibers (RCF)in myelofibrosis (MF) [22] as a secondary event in myeloproliferative neoplasms (MPN) |
| Normal RF-0 |
N MF 0 |
No reticulin fibers, occasional individual fibers or focal areas with tiny amount of reticulin fiber network |
| Slight increase RF 1 |
+ MF 0 |
Fine reticulin fiber network throughout much of section and no course reticulin fibers |
| Moderate increase RF 2 | + + MF 1 |
Diffuse fine reticuline network with focal collections of thick course reticulin fibers and no collagenisation |
| Marked increase BM dry tap RF 3 |
+++ RCF = MF 2 |
Diffuse and dense increase in reticulin with extensive intersections, and presence of collagen fibers and no or minor osteosclerosis |
| OS Dry tap RF 4 |
Sclerotic RCF&O = MF 3 |
Diffuse and dense reticulin with coarse bundles of collagen associated with significant osteosclerosis (O) |
| Table 1: The 1980 Rotterdam Clinical and Pathological (RCP) criteria for Essential Thrombocythemia (ET) and Polycythemia Vera (PV)[1] | ||
| Clinical and molecular criteria | Bone marrow pathology criteria (WHO) |
|---|---|
| ET | Normocellular ET |
| 1. Platelet count of >350 x109/l and the presence of large platelets in a blood smear 2. Presence of JAK2-V617 F mutation 3. Normal erythrocytes, haemoglobin (Hb) and hematocrit (ht) |
Predominant proliferation of enlargedmature megakaryocytes with hyperlobulated nuclei and mature cytoplasm, lacking conspicuous morphological abnormalities. No increase, proliferation or immaturity of granulopoiesis or erythropoiesis. Reticuline fibrosis (RF) 0 or 1 |
| Prodromal PV | ET with features of PV |
| 1. Platelet count of > 350 x109/l and normal ht male <0.51, female <0.48, normal erythrocyte <5.8x1012/L is mandatory. 2. Presence of JAK2-V617F mutation 3. Low serum EPO level and/or increased LAP score 4. Spontaneous EEC. |
Increased cellularity with due to increased erythropoiesis or trilineage myeloproliferation (i.e. panmyelosis). Proliferation and clustering of small to giant (pleomorphic) megakaryocytes. Absence bone marrow features consistent with congenital polycythemia and secondary erythrocytosis. RF 0 or 1 |
| Prefibrotic hypercellular ET | ET.MGM |
| 1. Platelet count of >350 x109/l, 2. No signs of leuko-erythroblastosis 3. Slight or moderate splenomegaly on ultrasound 4. No anemia with hb and ht in the normal low normal range: hb>13g/dl 5. Presence of JAK2-V617 F mutation 6. No preceding or allied CML, PV, RARS-T or MDS. ------------------------------------------------- ET stage 4, borderline anemia and LDH↑ ET stage 5 Hb<12 g/dL, LDH↑↑, CD34+ ------------------------------------------------- ET stage 6, Post-ET MF (2008 WHO) |
Hypercellular ET due to chronic megakaryocytic and granulocytic myeloproliferation (ET.MGM) and normal or reduced erythroid precursors. Loose to dense clustering of more pleiomorphic megakaryocytes with hyperploid or clumpsy nuclei (not or some cloud-like). Prefibrotic: RF- 0/1, MF-0, no/minor splenomegaly -------------------------------------------------------- Early fibrotic ET:RF 2, MF 1, splenomegaly no/minor Fibrotic ET : RF3, RCF or MF2, overt splenomegaly ------------------------------------------------------------ Post-ET MF: RF3/4, or MF-2/3 (2008 WHO PMF) |
| Table 2: WHO and European clinical molecular pathological (WHO - ECMP) criteria for diagnosis and classification of prefibrotic JAK2V617F mutated prefibrotic ET [15,25] | |
| Clinical and molecular criteria | Bone marrow pathology criteria (WHO) |
|---|---|
| Major Prodromal PV (ET stage 2). Hematocrit upper limit of normal: Ht: 0.45 to 0.51 in male and 0.43 to 0.48 in female), erythrocytes <5.8x1012/L PV A1. Hematocrit >0.51/>0.48 in male/female, erythrocytes >5.8x1012/L A2. Presence of heterozygous or homozygous JAK2V617F or JAK2 exon 12 mutation A3. Low serum Epo level Minor B1. Persistent increase of platelet count: grade I: 400-1500, grade II: >1500 x109/L B2. Granulocytes >10 x109/l or Leukocytes >12 x109/l and/or raised LAP-score in the absence of fever or infection B3. Splenomegaly on palpation or on ultrasound echogram (>12 cm length in diameter). B4. Spontaneous endogenous erythroid colony (EEC) formation (optional) |
PV. Bone marrow pathology: increased cellularity (60-100%) due to trilinearincrease of erythropoiesis, megakaryopoiesis and granulopoiesis and clustering of small to giant (pleomorph) megakaryocytes with hyperlobulated nuclei. Absence of stainable iron. No pronounced inflammatory reaction (plasmacytosis, cellular debris) Erythrocytosis. Increase of erythropoiesis, normal granulopoiesis and megakaryocytes of normal size, morphology and no clustering in primary/secondary erythrocytosis. Grading of reticulin fibrosis (RF) and myelofibrosis (MF) Erythrocythemic stage: A1 and P1 Prefibrotic: RF-0/1 = MF-0 Early fibrotic: RF-2 = MF-1 Fibrotic: RCF 3 = MF-2 Post-PV MF: RF 4 = MF-3 |
| On top of WHO bone marrow histology: ECMP criteria for staging of early, overt, and advanced PV A0, A2, B1 establish early PV (mimicking ET) PV ECMP stage 0, or masked PV A1, A2, and none of B establish polycythemic PV ECMP stage 1 A1, A2, and one or more of B establish classic and advanced PV ECMP stage 2 and 3 A2, B3 and P1 detect masked ET or PV: primary myeloproliferative disease (PMD) A3 and B4, bone marrow smear and JAK2V617F mutation is an important research option. Table 3: WHO-ECMP criteria for the diagnosis of PV and erythrocytoses [15,25] |
|
| Clinical and molecular (CM) JAK2 wild type ET | Bone marrow pathology (P) criteria (WHO) |
|---|---|
| 1. Platelet count >350x109/L and presence of large platelets in blood smear 2. Hemoglobin, haematocrit and erythrocyte count in the normal range 3. Presence of MPL515 mutation and JAK2 wild type 4. Normal serum EPO 5. Normal LAP score and CD11b expression 6. No or slight splenomegaly 7. No leukoerythroblastosis 8. No preceding or allied CML, PV, RAS-T or MDS |
P1. Proliferation of large to giant mature megakaryocyte with hyperlobulated, staghorn-like nuclei in a normocellular bone marrow (<65%) No increase of erythropoiesis, and no increase or immaturity of granulopoiesis or erythropoiesis, No or slight increase in reticulin RF 0/1 ET → MF Increased reticulin fibrosis around dense clustered megakaryocytes in a normocellular bone marrow and reduced erythropoiesis. Follow-up data of RF and MF related to splenomegaly in MPL515 ET transltional states to MF are lacking. Grading of reticulin fibrosis (RF) and myelofibrosis (MF) is similar as described for PV |
| Table 4: 2014 WHO Clinical Molecular and Pathological (2014 WHO-CMP) criteria for the diagnosis of normocelular ET carrying one of the MPL515 mutations [15,25]. This entity is identical to 'true' ET as defined in 2002 by Michiels & Thiele [5]. | |
| Clinical (CM) criteria JAK2 wild type PMGM | Pathological ECP criteria of CALR MGM |
|---|---|
| A1 No preceding or allied other subtype of myeloproliferative neoplasm PV, CML, MDS. The main presenting features is pronounced isolated thrombocythemia with platelet count around or above 1000x109/L A2 Presence of CALR mutation and JAK2 wild type C Clinical stages of CALR MGM C1. Early clinical stage: Hb>12g/dL, slight to moderate splenomegaly, thrombocytosis around or above 1000x109/L, normal LAP score C2. Intermediate clinical stage: slight anemia Hb<12 to >10 g/dL, decreasing platelet count, splenomegaly, increased LDH and definitive tear drop erythrocytes C3. Advanced stage: anemia Hb<10g/dL, tear drop erythrocytes, increased LDH, increased CD34+ cells, pronounced splenomegaly, normal or decreased platelet counts, leucocytosis or leukopenia. |
P1 Primary megakaryocytic granulocytic myeloproliferation (PMGM) and relative or absolute reduction of erythropoiesis and erythroid precursors. Abnormal dense clustering and increase in atypical medium sized, large to giant immature megakaryocytes containing bulbous (cloud-like) hypolobulated nuclei and definitive maturation defects
|
| The combination of A1 + A2 and P1 establishes CALR ET and various clinical stages (C1, C2,C3) related to the degree of myelofibosis (MF) Table 5: 2016 WHO-CMP criteria for hypercellular ET associated with primary megakaryocytic, granulocytic myeloproliferation (PMGM) caused by calreticulin (CALR) mutations [15,25]. This entity has been defined as PMGM in the 1990 Bone Marrow Classification by Georgii, et al. [22] as the third distinct MPD entity. |
|
| Number | Leukoc | LAF | Ery/Plasm | BM | BM | BM | Platelets | Hb | Ht | Erythroc |
|---|---|---|---|---|---|---|---|---|---|---|
| 109/L | score | volume | RF | megakar | cellularity | 109/L | mmol/l | 1012 /L | ||
| A1 ETPV | 10 | 183 | 31 / 41 | N | 1+ | N | 792 | 10.4 | 0.49 | 6.7 |
| A2 ETPV | 9 | 155 | N | 1+ | N | 887 | 10.0 | 0.51 | 6.0 | |
| A3 ET | 8 | 109 | 1+ | 2+ | 2+ | 911 | 8.9 | 0.47 | 5.4 | |
| A4 ET | 9 | 101 | 1+ | 614 | 8.0 | 0.39 | 4.5 | |||
| A5 ET | 16 | 128 | 26 / 37 | N | 1+ | N | 939 | 8.3 | 0.40 | 4.4 |
| A6 ET | 7 | 139 | N | 1+ | N | 742 | 9.8 | 0.49 | 5.8 | |
| A7 ET | 8 | 127 | N | 1+ | N | 567 | 9.5 | 0.46 | 5.2 | |
| A8 ET | 10 | 38 | 1+ | 2+ | 1+ | 875 | 8.8 | 0.43 | 4.9 | |
| A9 ET | 10 | 103 | 1+ | 1+ | 1+ | 690 | 8.8 | 0.45 | 5.5 | |
| A10 ET | 11 | 60 | N | 1+ | N | 1440 | 8.6 | 0.43 | 4.7 | |
| A11 ETPV | 13 | 113 | 32 / 43 | 2+ | 2+ | 1+ | 1435 | 9.4 | 0.46 | 6.1 |
| A12 PV | 10 | 207 | 59 / 52 | 1+ | 1+ | 2+ | 1932 | 11.1 | 0.56 | 6.5 |
| A13 PV | 28 | 193 | N | 2+ | 2+ | 1800 | 12.1 | 0.62 | 7.6 | |
| A14 PVT | 13 | 236 | 2+ | 2+ | 2+ | 952 | 8.3 | 0.45 | 5.6 | |
| A15 PVT | 11 | 103 | 1+ | 2+ | N | 636 | 7.7 | 0.39 | 5.4 | |
| A16 PV | 17 | 243 | 45 / 38 | 1+ | 2+ | 2+ | 1065 | 13.4 | 0.68 | 7.9 |
| A17 PV | 8 | 186 | 60 / 51 | 1+ | 1+ | 1+ | 728 | 10.9 | 0.57 | 7.5 |
| A18 PV | 14 | 184 | 63 / 50 | 1+ | 2+ | 1+ | 1035 | 12.2 | 0.64 | 7.1 |
| A19 PV | 16 | 219 | 50 / 40 | 1+ | 2+ | 2+ | 1320 | 13.3 | 0.70 | 6.4 |
| A20 PV | 18 | 128 | 38 / 52 | 1+ | 2+ | 1+ | 1300 | 11.9 | 0.65 | 7.6 |
| A21 PV | 13 | 170 | 43 / 37 | 2+ | 2+ | 2+ | 1085 | 12.1 | 0.61 | 7.1 |
| A22 PV | 17 | 168 | 42 / 35 | 1+ | 2+ | 2+ | 708 | 11.0 | 0.59 | 7.5 |
| A23 PV | 9 | 219 | 54 / 42 | 1+ | 2+ | 2+ | 959 | 13.1 | 0.72 | 9.1 |
| A24 PV | 18 | 215 | 82 / 46 | 2+ | 609 | 12.5 | 0.66 | 9.9 | ||
| B 1 PV | 235 | 38 / 36 | 2+ | 2+ | 2+ | 2975 | 5.3 | 0.32 | 4.4 | |
| B 2 ET | 5 | 20 | 28 / 58 | 1+ | 2+ | 699 | 8.3 | 0.42 | 4.0 | |
| B 3 PV | 14 | 140 | 58 / 62 | 1+ | 2+ | 2+ | 918 | 11.3 | 0.38 | 7.3 |
| B 4 PV | 23 | 140 | 44 / 51 | 2+ | 2+ | 2+ | 2500 | 12.7 | 0.63 | 7.7 |
| B 5 ET | 18 | 118 | 30 / 38 | 1+ | 1+ | N | 810 | 10.0 | 0.50 | 5.1 |
| B 6 ET | 9 | 123 | 28 / 37 | 1+ | 1+ | N | 737 | 9.0 | 0.47 | 5.0 |
| A= complicated by erythromelalgia and/or migraine-like atypical transient ischemic attacks, B= asymptomatic ET or PV, ET= essential thrombocythemia, PV= polycythemia Vera, PVT=PV after phlebotomy Table 6: Peripheral blood and bone marrow data in 14 ET and 16 PV patients observed between 1975 and 1981, University Hospital, Dijkzigt, Rotterdam [1] |
||||||||||
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Age, years. F /M | 56/M | 60/M | 66/F | 47/F | 40/F | 31/F | 50/M | 43/F | 47/F | 38/M |
| Platelets x109/L | 575 | 814 | 544 | 553 | 425 | 576 | 397 | 405 | 924 | 384 |
| JAK2V617F | +/- | +/- | +/- | +/- | +/- | +/- | ++ | +/- | ++ | ++ |
| Serum EPO | Normal | zero | decreased | decreasd. | nt. | decreased. | zero | decreased | zero | zero |
| Leukocytes x109/l | 6.7 | 5.3 | 12.9 | 8.2 | 6.1 | 6.2 | 7.3 | 14.3 | 13.1 | 8.0 |
| LAP score (N=<100) | 9nt | 160 | 197 | N | N | 186 | 163 | 263 | 232 | 284 |
| Hemoglobin g/dl | 13.6 | 15.5 | 14.2 | 14.4 | 13.4 | 14.0 | 18.6 | 17.3 | 16.3 | 12.8 |
| Hematocrit | 0.40 | 0.45 | 0.44 | 0.44 | 0.40 | 0.41 | 0.63 | 0.52 | 0.53 | 0.60 |
| Erythrocytes x1012/L | 4.5 | 5.3 | 4.7 | 4.8 | 4.6 | 4.9 | 6.3 | 6.1 | 7.4 | 6.7 |
| EEC | + | + | + | nt. | nt. | +. | + | + | + | + |
| Red cell mass | N | N | N | N | nt. | nt. | ↑ | ↑ | ↑ | ↑ |
| Spleen, echogram cm | . | 13 | 16 | 13 | 16.5 | 11.8 | 13.7 | 13 | 14.3 | 16 |
| Clinical diagnosisNo use of BM | ET | ET | ET | ET | ET | ET | PV | PV | PV | PV |
| Table 7A: Clinical features of 6 ET, and 4 patients carrying the JAK2V617F mutation | ||||||||||
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Cellularity | 60% | 65% | 90% | 75% | 80% | 75% | 80% | 65% | 80% | 80% |
| M:E ratio | 1 | 1 | 1 | 0,5 | 4 | 0,7 | 0,7 | 1 | 1.5 | - |
| Myeloid lineage | N | N | ↑ | N | ↑ | N | N | N | ↑ | ↑ |
| Erythroid lineage | ↑ | ↑ | ↑ | ↑ | N | ↑ | ↑ | ↑ | ↑ | ↑ |
| Myelofibrosis: MF | MF-0 | MF-1 | MF-0 | MF-0 | MF-0 | MF-0 | MF-0 | MF-0 | MF-1 | MF-0 |
| Diagnosis 2008 WHO | ET | ET | ET | ET | MPNuc | ET | PV | PV | PV | PV |
| Bone marrow diagn | ET | ET | PV | PV | EMGM | PV | PV | PV | PV | PV |
| Diagnosis2008 ECMP | ET1 | ET1 | Pro-PV | Pro-PV | EMGM | Pro-PV | PV | PV | PV | PV |
| Follow-up up to 2008 | 4 yrs | 12 yrs | 10 yrs | 11 yrs | 15 yrs | 8 yrs | 4 yrs | 1 yrs | 5 yrs | 1 yrs |
| Treatment 2008 | aspirin | aspirin | Low IFN | HU | no | High IFN | Phleb | Phleb | Phleb/HU | Phleb/HU |
| Diagnosis in 2012 | ET | ET | Remission | PV | EMGM | PV | PV | PV | PV | PV |
| Treatment 2015 | aspirin | aspirin | Very low IFN | HU | no | Pheb | Phleb | Phleb | HU | HU |
| Table 7B: Bone marrow features of 10 JAK2V617F mutated thrombocythemia patients with the clinical diagnosis of ET in 6 and PV in 4 cases | ||||||||||
| 1971 | 1978 | 1985 | 1988 | 1989 | 1989a | |
|---|---|---|---|---|---|---|
| Erythromelalgia | - | - | + | + | + | - |
| Peripheral blood | ||||||
| Hemoglobin mmol/l | 9.8 | 8.0 | 7.1 | 5.9 | 5.7 | 5.8 |
| Platelets X109/l | 285 | 265 | 421 | 484 | 811 | 223 |
| LeukocytesX109/l | 6.9 | 10.8 | 21.4 | 21.9 | 24 | 18 |
| Bone Marrow | ||||||
| Cellularity | N | ↑ | ↑ | ↑ | ||
| Erythropoiesis | N | N | N | N | ||
| Ringed sideroblasts | - | - | - | - | ||
| Megakaryocytes | ↑ | ↑ | ↑↑ | ↑↑ | ||
| Reticulum | ↑ | ↑↑ | ↑↑↑ | ↑↑↑ | ||
| Collagen | - | - | ↑ | ↑↑ | ||
| Osteoid | - | - | - | + | ||
| Myelodysplasia | - | - | - | - | ||
| Karyotype of bone marrow cells | ||||||
| Spleen size on scan | ||||||
| Length diameter cm | 18 | 23 | ||||
| *N, normal ; ↓,decreased; ↑, increased; -, absent; +, present. One arrow slight, two arrows pronounced, and three exceptional. The peripheral blood and bone marrow findings are consistent with agnogenic myeloid metaplasia in case 1.a While on treatment with Hydroxyurea Source Michiels and Ten Kate, Amer J Hematol 1992; 39: 131-136 [17] Table 8: Hematological Findings in Cases of Erythromelalgia and Atypical Thrombocythemia* |
||||||




































