Open Access
Case Report
Max Screen >>
ISSN: 2348-9820
Copyright: © 2016 Koch K. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
A 49-year-old man presented with a 2-year history of an enlarging ulcerated plaque over the right forearm with an associated history of pruritus. Skin biopsy demonstrated an infiltrate of lymphocytes in the dermis and subcutis, together with atypical cells with large nuclei and prominent nucleoli resembling Reed-Sternberg cells. Immunohistochemical staining showed that the cells were positive for CD30 and CD15, but negative for LCA, CD20, CD3 & ALK1, consistent with classic Hodgkin Lymphoma. Computerized tomography, bone marrow biopsy and blood work-up revealed no systemic features, confirming a diagnosis of Primary Cutaneous Hodgkin lymphoma (PCHL). The patient received four cycles of ABVD (adriamycin, bleomycin, vinblastine and dacarbazine) with regression of the tumor but subsequently died due to pulmonary tuberculosis and pulmonary thromboembolism.
Keywords: Primary Cutaneous Hodgkin lymphoma; PCHL; Hodgkin lymphoma; HL; Cutaneous lymphoma
List of Abbreviations: HL: Hodgkin Lymphoma; PCHL: Primary Cutaneous Hodgkin Lymphoma; CHL: Cutaneous Hodgkin Lymphoma; MOPP: Mechlorethamine, vincristine, Procarbazine, Prednisone; ABVD: Adriamycin, Bleomycin, Vinblastine and Dacarbazine; RT: Radiation Treatment; CT: Corticosteroids; EBV: Epstein-Barr virus; HIV: Human Immunodeficiency Virus; LyP: Lymphomatoid Papulosis; ALCL: Anaplastic Large Cell Lymphoma
Whilst the association of Hodgkin Lymphoma (HL) with skin involvement is well described in the literature, Primary Cutaneous Hodgkin Lymphoma (PCHL) is exceedingly rare, with less than 10 cases reported to date in the literature. PCHL occurs when Cutaneous Hodgkin Lymphoma (CHL) arises in the skin without evidence of systemic disease. Immunohistochemistry has allowed for more accurate diagnosis of this condition, leading to speculation of PCHL reported prior to the 1990s. The prognosis for PCHL is variable. We share our experience in diagnosis and treatment of this condition.
A 49-year old man presented with a 2-year history of a gradually enlarging 10cm isolated, ulcerated plaque over the right forearm (Figure 1). He had associated pruritus but was constitutionally well with no symptoms of fever, weight loss or night sweats. He had no pallor or jaundice. A single right axillary lymph node measuring 1 by 2 cm in diameter was found with no widespread lymphadenopathy or hepatosplenomegaly. He had no symptoms of tuberculosis on history or possible tuberculosis contacts.
A 3mm skin punch biopsy of the right forearm was submitted for histopathological assessment. Histology showed an infiltrate of polymorphous lymphocytes, neutrophils and plasma cells in the dermis and subcutis. Importantly, there were intermixed atypical cells displaying binucleation, large nuclei and prominent nucleoli, resembling Reed-Sternberg cells (Figure 2a). Immunohistochemical staining showed that the cells were positive for CD30 and CD15 (within membranous and paranuclear regions) and negative for LCA, CD20, PAX5, CD3, EBV-LMP1, ALK1 & EMA (Figure 2b and c). EBV in situ hybridization was negative. These findings were in keeping with cutaneous infiltration by classic HL.
Excision biopsy of the right axillary node revealed reactive follicular hyperplasia without any evidence of HL.
Laboratory investigations (full blood count, renal function, liver function, lactate dehydrogenase and erythrocyte sedimentation rate) were normal. Epstein-Barr virus (EBV) and Human immunodeficiency virus (HIV) serology were negative. A staging computerized tomographic (CT) scan and bone-marrow trephine biopsy did not reveal evidence of systemic involvement by Hodgkin Lymphoma (HL). The above investigations confirmed that our patient had PCHL.
He was treated with four cycles of ABVD (adriamycin, bleomycin, vinblastine and dacarbazine) over a period of 4 months. The cutaneous lesion responded to treatment with complete shrinkage of the plaque (Figure 3). However, the patient developed pulmonary tuberculosis and pulmonary thromboembolic disease shortly after the fourth cycle of chemotherapy and died.
Hodgkin lymphoma (HL) primarily affects the lymph nodes. Cutaneous involvement is rare but well described. It typically occurs in late stage disease and carries a poor prognosis [1]. Cutaneous involvement by HL can be divided into specific and non-specific subtypes. Specific cutaneous HL, where malignant cells infiltrate the skin was first described by the German Physician Grosz in 1906 [2]. Nonspecific HL may present as a paraneoplastic syndrome with pruritus, erythroderma, ichthyosis, dermatomyositis, urticarial, eczematoid or psoriatic eruptions with no direct tumor spread to the skin [3].
Skin involvement is found in HL in 0.5-3.4% of cases [4,5]. Due to the rarity of PCHL its existence remained a matter of debate until 1994 when Sioutos et al. published the first verifiable series of cases with a diagnosis based on immunohistochemistry and modern imaging techniques [6]. Retrospectively, it is thought that cases published by Van der Meiren (1948) [7] and Szur (1970) [8] can be better classified as other disease entities.
Morphologically similar lymphomas such as lymphomatoid papulosis (LyP), granulomatous skin lymphoma, anaplastic large cell lymphoma (ALCL) and regressing atypical histiocytosis [1] would need to be considered in the differencial diagnosis [9].
The diagnosis of PCHL is made when a patient presents with Hodgkin Lymphoma with no systemic involvement. It is possible that PCHL may represent spread from an occult primary nodal HL [10]. Spread of nodal HL to the skin is thought to occur via direct extension from overlying skin, retrograde lymphatic spread or haematogenous spread [4]. In our case, an ipsilateral axillary lymph node was found, but excision and histological examination revealed dermatopathic changes, without evidence of HL.
Lesions of PCHL have been described as erythematous papules and nodules with or without ulceration. Two patients have been reported to have had background eczematous plaques similar to our patient [6,7]. In the majority of reported cases, PCHL affects the extremities [1,3,6-8,10].
There is no standard of care treatment for PCHL. Treatment varies and is generally decided on an individual ‘case-by-case’ basis. Treatment includes topical agents, systemic combination chemotherapy and radiotherapy. In the report by Sioutos et al. (1994), two patients who received only topical corticosteroids had excellent outcomes. One patient received local radiation therapy with a good local response followed by systemic progression at six years. Two patients were treated with systemic chemotherapy - MOPP-ABVD, given as six cycles. This resulted in complete remission in one patient (with follow up at five years) and progression in the second patient (at two months) [6]. Mukesh et al. (2009) treated their patient with six cycles of ABVD with no progression at three-year follow up [1]. Guitart et al. (1998) treated a patient with local radiation therapy which resulted in a good cutaneous response, but systemic progression after one year [10].
The outcome of PCHL is variable. Sioutos et al. (1994) reported two of their five patients developing HL lymph node involvement at two months and six years respectively, while they found no progression in three of their five patients at 5, 9 and 20 years follow-up [6]. Guitart et al. (1998), reported on a patient with PCHL who developed systemic lymphoma after one year and subsequently demised [10] (Table 1).
PCHL is an extremely rare entity. Immunohistochemical characterization is essential to making a diagnosis of isolated skin involvement. Despite the benign prognosis described in several reports, some patients do develop systemic (nodal) HL. Longterm follow-up is essential to detect progression to systemic HL in a subset of patients with PCHL.

![]()

 |
| Figure 1: Ulcerated plaque on the right forearm (a) and close up view |
   |
| Figure 2: (a) Reed Sternberg cells (haematoxylin & eosin) showing (b) CD15 and (c) CD 30 immunoexpression within membranous and paranuclear regions of tumour cells |
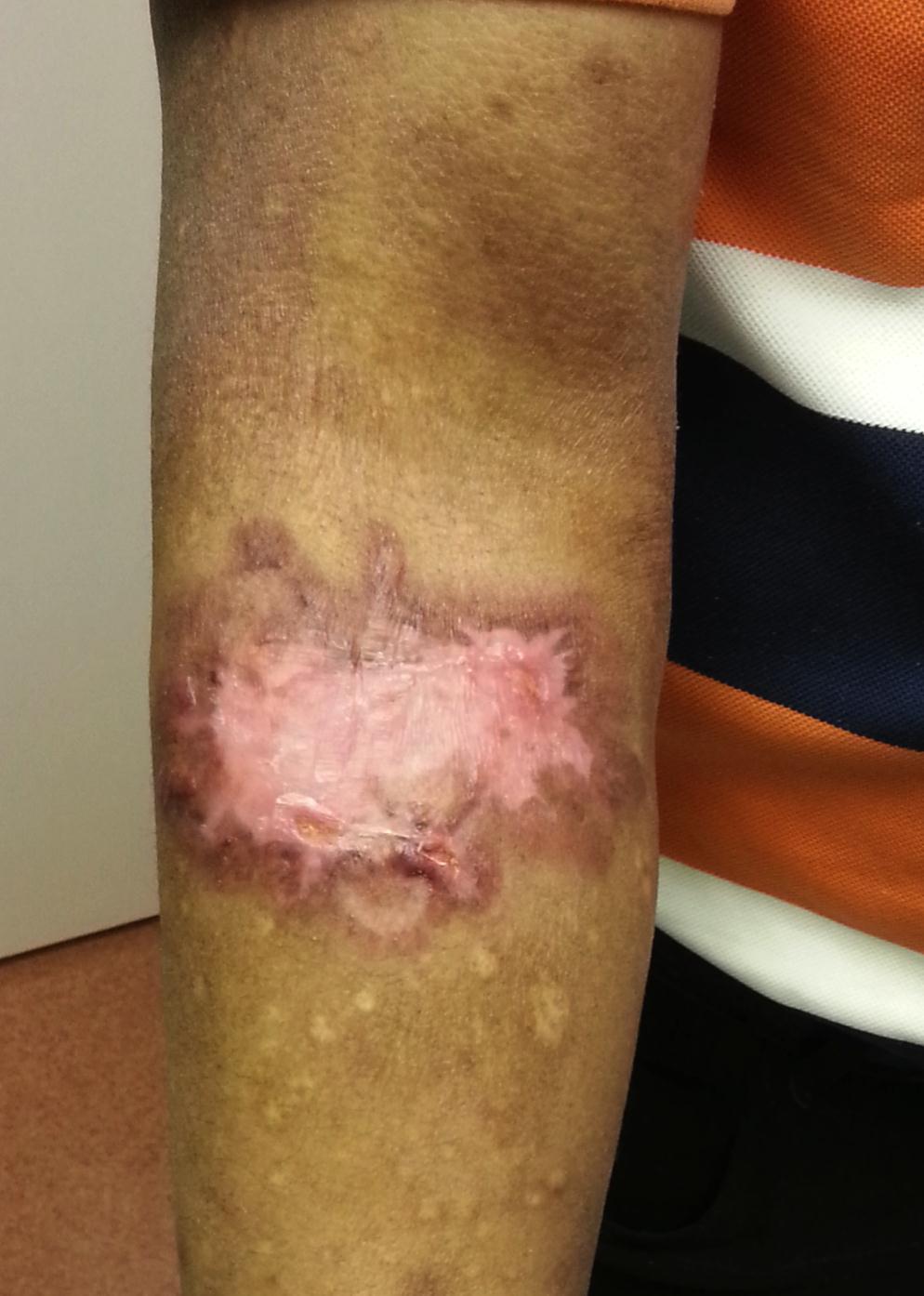 |
| Figure 3: Right forearm, following ABVD chemotherapy |
Authors |
Age |
Gender |
Symptoms |
Treatment |
Response |
|---|---|---|---|---|---|
Sioutous et al. (1994) [6] |
50 |
M |
Nodules on chest wall |
MOPP-ABVD |
Systemic disease after 2 months |
54 |
M |
Nodules on left leg |
RT |
Systemic disease after 6 years |
|
45 |
F |
Nodules on arms and legs |
Multiple chemotherapy |
No progression (5yrs) |
|
17 |
M |
Nummular eczema-like lesions, papules right thigh |
Topical CT |
No progression (9yrs) |
|
52 |
M |
Tumour nodules and papules on right arm |
None |
No progression (20yrs) |
|
Guitart et al. (1998) [10] |
86 |
M |
Ulcerated nodule left ankle |
RT |
Progression after 1 year |
Mukesh et al. (2009) [1] |
59 |
M |
Painless erythematous indurated lesions left flank, inner thigh and right foot |
ABVD |
No progression (3yrs) |
Pratenda et al. (2009) [3] |
70 |
M |
Erythematous papules and nodules on the back |
RT |
No progression (7yrs) |
| MOPP: Mechlorethamine, Vincristine, Procarbazine, Prednisone; ABVD: Adriamycin, Bleomycin, Vinblastine and Dacarbazine; RT: Radiation Treatment; CT: Corticosteroids Keyword search: Primary Cutaneous Hodgkin Lymphoma, Cutaneous Hodgkin Lymphoma, Hodgkin lymphoma involving the skin Table 1: Reported Primary Cutaneous Hodgkin Lymphoma |
|||||




































