Open Access
research article
Max Screen >>
ISSN: 2394-6504
Copyright: © 2015 Animasahun BA. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Background: Dilated Cardiomyopathy is the most common form of the cardiomyopathies in children accounting for a significant cause of morbidity and mortality as well as a common indication for heart transplant. Although dilated cardiomyopathy is a common indication for Cardiac transplantation, there are still treatable causes of dilated cardiomyopathy. We aim to document the cases seen in children in a tertiary Hospital in South-Western Nigeria over an eight year period, mode of treatment and their outcomes.
Methods: Prospective and cross sectional involving consecutive patients diagnosed with dilated cardiomyopathy using echocardiography at the Paediatric Department of Lagos State University Teaching Hospital, Lagos Nigeria as part of a large study between January 2007 and December 2014.
Results: Dilated Cardiomyopathy was diagnosed in 22 (1.55%) of children with structural heart disease with a male to female ratio of 1:1.2 with mean age±SD of 4.73±3.50. The most common mode of presentation was congestive cardiac failure which occurred in 54.55% of the study subjects. Ten of the subjects had no identifiable cause for the DCM. Five of the them had Tachycardia induced cardiomyopathy. All but one were treated medically with antifailure drugs and calcium, while only one patient had surgery. Almost half of the patients got well with the above treatment and did not require cardiac transplantation.
Conclusion: Rare causes of dilated cardiomyopathy such as tachycardia induced cardiomyopathy, anomalous origin of the left coronary artery arising from the pulmonary artery syndrome, Burns Cardiomyopathy and Adriamycin Induced Cardiomyopathy were documented in this study, all the patients with tarchycardia induced cardiomyopathy and burns cardiomyopathy recovered with medical treatment only.
Keywords: Cardiomyopathy; Children; Cardiac failure; Cardiac transplantationDilated cardiomyopathy (DCM) is a primary myocardial disease characterized by varying degrees of left ventricular dysfunction and dilatation in the absence of chronic increased afterload or volume overload [1]. Rarely right ventricular dysfunction may occur. DCM is the most common form of the cardiomyopathies in children accounting for a significant cause of morbidity and mortality as well as a common indication for heart transplant [2,3].
It is most commonly diagnosed in younger children with an average age of two years in developed countries. The incidence of DCM in developed countries is 0.57–1.23 per 100,000 per year in children younger than 18 years [4,5]. In the study in America, the incidence was higher in boys than in girls, in blacks than in whites and in infants compared to older children [5]. In African children, DCM is also more common in children less than five years [6,7], however the incidence is scarcely defined.
There are diverse causes of DCM but it is largely unknown or idiopathic in most cases. It can be primary (genetic, mixed or predominantly familial non-genetic or acquired) or secondary (infiltrative or autoimmune) [8]. It has varied genetic abnormalities with mutations in up to 40 genes identified [9-12]. The underlying genetic causes of 50-60% of DCM cases are unknown [13]. In children, two thirds of the cases of DCM are idiopathic and the most common cause is myocarditis and neuromuscular disease [13].
Apart from Genetic causes of DCM in children can be a result of infection, inflammation, endocrine or metabolic disorders or malnutrition [14-16]. Infants and children have a wider spectrum of causes [17-20] and identifying the aetiology may be challenging especially in resource poor and developing countries. Other causes in children include long standing supraventricular tachycardia, burns cardiomyopathy, anomalous origin of the left coronary artery arising from the pulmonary artery and critical coarctation of the aorta [21-24].
In resource poor countries where facilities may be unavailable for genetic and more advance investigations, the clinical presentation with a thorough evaluation of the patients will define the possible aetiology.
Although DCM is a common indication for Cardiac transplantation, there are still treatable causes of dilated cardiomyopathy such as those secondary to anomalous origin of the left coronary artery from the pulmonary artery(ALCAPA), Coarctation of the aorta, Arrhythmia and carnithine anhydrase defficiency, however our undocumented observation in the region is that a diagnosis of dilated cardiomyopathy is almost synonymous with death penalty due to unavailability of cardiac transplantation services in the region.
There are very few studies on DCM in children in Nigeria. To the best of the authors awareness, the only study which described the profile of patient children with DCM was documented by Bode Thomas, et al. [7] among children children in Jos Northern part of Nigeria. There has been no data on children from the South-western part of the country. Only one study has also discussed the outcome in these patients hence the need for this study which aims to document the cases of DCM seen in children in a tertiary Hospital in South-Western Nigeria over an eight year period and their outcomes and compare those findings with reported cases within and outside the region, the data obtained will add to the very few available in the region.
This is a review of prospectively collected data of all patients diagnosed with dilated cardiomyopathy using echocardiography at the Paediatric Department of Lagos State University Teaching Hospital (LASUTH) Lagos Nigeria as part of a large study between January 2007 and December 2014.
LASUTH is a tertiary institution in Lagos, Southwestern Nigeria and receives referral from the region. The Estimated Population of Lagos alone in the 2006 census was 18 million, however LASUTH receives referral from Lagos and other neighbouring states like Oyo, Ogun and Osun hence serves a population of more than 30 million Nigerians altogether. The general Paediatric department ward is an 83 bed-ed ward. The out-born unit is equipped with 14 cots and 12 incubators. A Paediatric Cardiologist is in charge of the cardiology unit. Patients referred to the cardiology unit of the Department are evaluated with chest radiograph, electrocardiography and echocardiography and other ancillary investigations as required.
One echocardiography machine was used on all the subjects throughout the study period. The machine is a GE vivid Q echocardiography machine reference number 14502 WP SN 2084. It has facility for two dimensional, M-mode and color flow Doppler imaging [25,26].
The Paediatric Cardiologist performed the echocardiography on all the subjects. The diagnosis of Dilated cardiomyopathy was made based on standard definitions [1,12,13].
Ethical approval was obtained from the research and ethics committee of the Lagos State University Teaching Hospital while consent were obtained from the parent or caregivers before inclusion in the study.
The data were imputed into a computer and analyzed using Statistical Package for Social Sciences (SPSS) version 20. The children's age, sex, indication for echocardiograph and diagnosis, mode of treatment interms of surgical or medical and outcomes of the patients were documented. Tables and charts are used to depict those variables. Means of continuous variables were compared using the Student t test and proportions using Chi-square test. Level of significance set at p< 0.05.
A total of 1495 children ≤13 years had echocardiography done within the study period and 1126 (75.32%) had structural heart disease. DCM was the most common cardiomyopathy in the study subjects comprising 71% of the cardiomyopathies. DCM was diagnosed in 22 (1.55%) of children with structural heart disease. It was diagnosed in 12 females and 10 males with a male to female ratio of 1:1.2. The children were between nine weeks and 11 years (mean of 4.73±3.50) with a median age of 3.4 years. The mean ages of the males and females was 4.14±3.67 and 5.18±3.46 respectively. The difference in the mean age between males and females is not statistically significant (p=0.84). Twelve children were ≤5 years, eleven were ≥5 years and the age of one was not documented. The age distribution is illustrated in Figure 1.
All the patients were ill at presentation. The most common mode of presentation was congestive cardiac failure which occurred in 54.55% of the study subjects. Others presented with upper respiratory tract infection, generalized Oedema, tachycardia, palpitations, advanced retroviral disease. Table 1 shows the indication for echocardiography in the subjects.
In terms of the causes of DCM as shown in Table 2. Ten of the subjects had no identifiable cause for the DCM. Nine out of which presented in congestive cardiac failure without a preceding history of a suspected or confirmed heart lesion or a febrile illness. Those subjects comprised six females and three males with a mean age of 5.74±3.70. The tenth subject was a 7 year old male who presented with generalised oedema.
In the mode of treatment of the subjects; All but one were treated medically with antifailure drugs (diuretics, digoxin and ACE inhibitors), aspirin and calcium, while only one patient had surgery (re-implatation of the origin of the anomalous coronary artery to the aorta.
The outcome of the subject according to the causes of dilated cardiomyopathy are shown in Table 3.
Almost half of the patients got well with the above treatment and did not require cardiac transplantation.
The incidence of dilated cardiomyopathy recorded in children less than 13 years during the study period was 6.9 per 100,000 children who presented at the study centre during the period of study, it constituted 1.55% of those with structural heart diease but 17.6% of those with acquired heart diseases and 71% of those with cardiomyopathy, this is consistent with similar report within and outside Africa [4,27,28].
Altogether only 22 cases were diagnosed during the study period, this proportion appear small but is comparable to the number documented in other part of the country. The only earlier documentation on dilated cardiomyopathy in children in Nigeria were by Bode Thomas, et al. In 2005 [7] they documented only five cases over a period of two and half years, in the northern part of the country, In their later study in 2013, Bode-Thomas, et al. [27] also documented 33 cases over a period of 10 years.
An incidence of 6.9 per 100,000 children is actually higher than 0.57 per 100,000 documented by Towbin, et al. [5] in their multicentred and longitudinal study over a period of six years among American and Canadian Children.
The male to female ratio of DCM in the study subjects was 1:1.2. This trend is similar with findings documented by Bode-Thomas, et al. in Nigeria [27], however in an earlier study by the same author there were more females compared to males [7]. This may have been due to the fewer subjects in the earlier study compared with the later study. Studies outside Africa have also documented different results, Nugent, et al. [4] in Australia documented more females with DCM while Towbin, et al. [5], in the USA reported a higher incidence in males compared to females.
In this study, children with cardiomyopathy were aged 9 weeks to 11 years with a mean age of 4.96±3.53 and a median age of 3.14 years. Half of those children were less than 5 years of age hence childhood DCM is more predominant in younger children. Similar report was documented by Bode-Thomas, et al. [4] and is also in keeping with reports in other Ccountries [4-7,27,28]. Not surprisingly, congestive cardiac failure was the most common mode of presentation in the subjects with DCM accounting for more than 50% of the presentation. This is also consistent with findings from other researchers [5-7,28-30].
It was not possible to carry out genetic, viral and other investigations to postulate possible etiologies for the DCM in our subject, therefore the subjects without identifiable cause, within the confines of our limitation, were labeled as idiopathic DCM. This limitation was also encountered by Bode thomal, et al. [27] However, for those subjects with identifiable cause, aetiologies identified included Tachycardia Induced Cardiomyopathy, Burns Cardiomyopathy, Viral Myocarditis, ALCAPA syndrome, Adriamycin Cardiomyopathy and Cardiomyopathy following HIV infection.
Viral aetiology has been implicated as a cause for majority of the cases of Cardiomyopathy [28,31,32]. Two of the study subjects had a prior upper respiratory tract infection (URTI) preceding the development of DCM, thus it may be possible that it is a viral infection with a myocarditis resulting in DCM. In the same vain, HIV has been recognized as an important aetiology for DCM [33-35]. Cases with advanced HIV, reduced CD4 counts and Zidovudine use are associated with increased risk of having DCM [36]. Our study subject was a six years old female with advanced HIV and PTB who subsequently developed DCM.
Prolonged tachycardia is a known cause of DCM [11]. It has been reported in infants and older children [37]. Five of the study subjects had Tachycardia induced cardiomyopathy, two were less than 2 years while two were over 5 years. Wenle, et al. [38] noted that more than half of their study subjects were infants and toddlers and 20% of the cases were adolescents [38]. To the best of the authors' knowledge there are no reported cases of childhood TIC in African children and this may be attributed to challenges with making this diagnosis in children and probably because it is an under-recognized cause of DCM and heart failure in children [39]. Adriamycin use is an established cause of DCM [40-42]. Although the incidence of this is not very common, we identified a case in the present study.
Cardiomyopathy has been reported following burns. The mechanism of action is poorly understood but it usually occurs days to weeks following severe burns and it has been reported in adults and children alike [43,44]. We documented in this paper an 8years old male with severe full thickness burns with a turbulent clinical course who presented weeks later with a congestive cardiac failure and DCM was diagnosed in him.
ALCAPA, anomalous origin of the left coronary arising from the pulmonary artery, syndrome is a rare but serious congenital anomaly [23]. It was first describe over a century ago [45]. Affected individuals present with signs and symptoms of heart failure in infancy or early childhood and left untreated has a mortality of up to 90% in infancy [46-48]. Two of our study subjects had ALCAPA syndrome and unsurprisingly, both presented in infancy with heart failure and breathlessness.
One of the major aim of this study was to document the treatable causes of dilated cardiomyopathy which does not require cardiac transplantation in view of the lack of this facility in our environment. Almost half of our patients (45%) had this which included tarchycardia induced cardiomyopathy, burns cardiomyopathy, ALCAPA, viral myocarditis, with 80% of them achieving complete recovery with antifailur drugs and calcium alone. Previous reports on dilated cardiomyopathy in the region [7,27,29]. did not include any of the above which we attribute to the rarity of these condition and possibly the difficulty in diagnosing these conditions. None of our subjects had dilated cardiomyopathy secondary to coartation of the aorta. One of the subjects with ALCAPA had surgery outside our region but died in the immediate post operative period. Our inability to carry out genetic studies and full metabolic studies could have also made us miss some of the cases of dilated cardiomyopathy which may be secondary to Carnithine deffieciency and other metabolic causes like hypothyroidism and might have been among those grouped as idiopathic, carnithine is also not available in our environment hence was not part of our therapy for the patients treated, availability of this might have saved some of those classified as idiopathic.
In Conclusion, rare causes of DCM such as Tachycardia Induced cardiomyopathy, ALCAPA syndrome, Burns Cardiomyopathy and Adriamycin Induced Cardiomyopathy were recorded in this study, all the patients with tarchycardia induced cardiomyopathy and burns cardiomyopathy recovered with medical treatment only. Even though we currently do not have cardiac transplantaion facility in the region, some patient with dilated cardiopathy can still be salvaged.

![]()

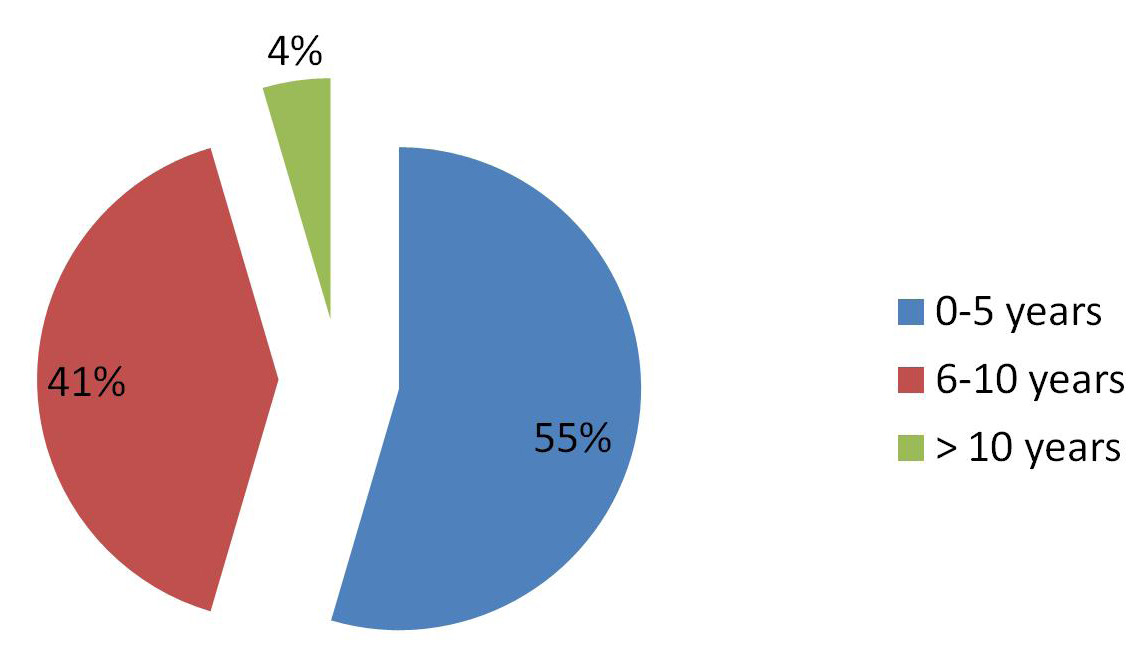 |
| Figure 1: Age categories of subjects |
| Indication | Frequency | Percent |
|---|---|---|
| CCF | 14 | 63.4 |
| RTI | 2 | 9.1 |
| Palpitation and CCF | 2 | 9.1 |
| suspected Myocarditis | 2 | 9.1 |
| CCF while on Chemotherapy | 1 | 4.5 |
| Advanced RVD | 1 | 4.5 |
| Total | 22 | 100 |
| RVD= retroviral disease, ALCAPA= anomalous origin of the left coronary artery from the pulmonary artery CCF= congestive cardiac failure, RTI= Respiratory tract infection Table 1: Indication for echocardiography in the subjects |
||
| Aetiology | Frequency | Percent |
|---|---|---|
| Idiopathic | 10 | 45.5 |
| Tarchycardia | 5 | 22.73 |
| ALCAPA | 2 | 9.1 |
| Burns | 1 | 4.5 |
| Myocarditis | 2 | 9.1 |
| Chemotherapy | 1 | 4.5 |
| Advanced RVD | 1 | 4.5 |
| Total | 22 | 100 |
| RVD= retroviral disease, ALCAPA= anomalous origin of the left coronary artery from the pulmonary artery Table 2: Aetiology of Dilated Cardiomyopathy in the subjects |
||
| Aetiology | Frequency | Alive | Dead or lost to follow up | p |
|---|---|---|---|---|
| Idiopathic | 10 | 3 | 7 | 0.07 |
| Tarchycardia | 5 | 5 | 0 | 0.002 |
| ALCAPA | 2 | 0 | 2 | 0.05 |
| Burns | 1 | 1 | 0 | 0.15 |
| Myocarditis | 2 | 1 | 1 | 0.15 |
| Chemotherapy | 1 | 0 | 1 | 0.15 |
| Advanced RVD | 1 | 0 | 1 | 0.15 |
| Total | 22 | 10 | 12 | 0.55 |
| RVD= retroviral disease, ALCAPA= anomalous origin of the left coronary artery from the pulmonary artery Table 3: Aetiology of Dilated Cardiomyopathy and outcomes in the subjects |
||||




































