Open Access
RESEARCH ARTICLE
Max Screen >>
ISSN: 2638-082X
Copyright: © 2015 Wylie MA. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
The C28/I2 line of immortalized juvenile human chondrocytes was employed to determine the extent to which recombinant human (rh) interleukin-6 (rhIL-6), the interleukin-6-like cytokine, rh-oncostatin M, and the adipokine, rh-adiponectin, activated extracellular-signal regulated kinase (ERK1/2), p38α mitogen-activated protein kinase (p38α MAPK) and c-Jun-amino-terminal kinase (JNK). Normal human chondrocytes incubated with rhIL-6 were also analyzed for JNK activation. Western blots which were quantified by the Metamorph® software program showed that p38α MAPK and JNK1/2, but not ERK1/2 were constitutively phosphorylated in C28/I2 chondrocytes. JNK1/2 was constitutively phosphorylated in normal human chondrocytes as well. C28/I2 chondrocytes produced primarily U-p42ERK (U-ERK2) where the ratio of phosphorylated (P) p42ERK to β-actin was significantly increased by rh-oncostatin M and rhIL-6, but not by rh-adiponectin. C28/I2 chondrocytes produced one form of unphosphorylated p38α MAPK. However in addition to P-p38α MAPK another form of P-p38α MAPK migrating at approximately 55kDa was also detected. Two forms of JNK migrating at approximately 51kDa and 46kDa which corresponded to JNK2 (p54) and JNK1 (p46), respectively, were produced by C28/I2 chondrocytes and normal human chondrocytes but an additional form of JNK (approximately 36kDa) was also found in C28/I2 chondrocytes. Although rhIL-6, rhOSM and rhAPN significantly reduced the content of U-JNK51kDa and U-JNK46kDa, none of these cytokines significantly activated JNK51kDa or JNK46kDa. Overall, these results implicated rhIL-6, and rh-oncostatin M in activating p42ERK by C28/I2 chondrocytes. These results may be helpful in defining genes regulated by IL-6-type cytokines in these cells.
Keywords: Chondrocytes; IL-6-type cytokines; Gene's regulation; Rheumatoid arthritis
List of Abbreviations: rhAPN: Recombinant human adiponectin; rhTNF-α: Recombinant human tumor necrosis factor-α; rhIL-6: Recombinant human interleukin-6; rhOSM: Recombinant human oncostatin M; ERK1/2: Extracellular Signal-Regulated Kinase1/2; JNK: c-Jun-NH2-terminal Kinase; MAPK: Mitogen-Activated Protein Kinase; MEK1/2: Mitogen-Activated Protein Kinase Kinase1/2; MMP: Matrix metalloproteinase; STAT: Signal Transducers and Activators of Transcription
Rheumatoid arthritis (RA) is a debilitating chronic inflammatory disease of synovial joints which over time ultimately alters the structure of articular cartilage and subchondral bone resulting in joint destruction. Over the past 15 years or so different clinical approaches to the treatment of RA with a pro-inflammatory cytokine receptor antagonist fusion protein, small molecule inhibitors, and anti-cytokine receptor monoclonal antibodies have been employed. These drugs were developed by identifying molecular targets which are key mediators in the pathogenesis and progression of RA [1-4]. However, there is much more to be learned about the signal transduction pathways involved in the initiation and progression of RA, especially how dysfunctional signal transduction alters articular chondrocyte homeostasis [5,6]. A further analysis of signal transduction may also reveal potential novel molecular targets for pharmacological intervention in RA necessary for inducing apoptosis of activated T- and B-cells, dendritic cells, neutrophils, macrophages and synoviocytes, while minimizing or inhibiting chondrocyte and osteocyte apoptosis [6-9].
Pro-inflammatory cytokines, most notably, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6 and IL-17 play significant roles in RA by activating various signal transduction pathways, including, the Stress-Activated Protein Kinase/Mitogen Activated Protein Kinase (SAPK/MAPK) and Janus Kinase/Signal Transducers and Activators of Transcription (JAK/STAT) pathways [10-12]. Deregulated activation of SAPK/MAPK and JAK/STAT signaling in inflamed RA synovial tissue [13-15] results in significant up-regulation of matrix metalloproteinases (MMPs), pro-inflammatory cytokines as well as anti-apoptosis proteins and pro-apoptosis proteins in RA articular cartilage [16].
Recent studies have indicated which pro-inflammatory cytokines germane to RA activate which signaling pathway; for example, IL-1β and TNF-α primarily targets SAPK/MAPK. In this regard, c-Jun-amino-terminal kinase (JNK) and p38 MAPK are activated by extracellular stressors and pro-inflammatory cytokines, including, IL-1β and TNF-α. These cytokines may also activate extracellular-signal regulated kinase (ERK1/2). Activation of ERK1/2 is critically important in the maintenance of chondrocyte homeostasis because ERK1/2 may alternately regulate cell proliferation and differentiation [17], depending on the specific extracellular signal engaged by its corresponding receptor tyrosine kinase. In contrast IL-6 and IL-17 primarily activates JAK/STAT [12]. Moreover, less evidence implicate IL-6 in activating articular chondrocyte SAPK/MAPK pathway. This gap in knowledge would be important to fill and thus be particularly relevant to RA because IL-6 and another member of the IL-6-type cytokines, namely oncostatin M (OSM) [18] as well as the adipokine, adiponectin (APN) [19] were found to be present at significantly elevated levels in RA synovial fluid [20] where they are conjectured to play a critical role in perpetuating chronic inflammation.
The principal objective of this study was to determine if recombinant human IL-6 (rhIL-6) and another cytokine related to IL-6, namely, rhOSM as well as the adipokine, rhAPN, the latter which interact with receptors distinctly different from the IL-6/IL-6R/gp130 complex, membrane IL-6R or soluble IL-6R, phosphorylate ERK1/2, JNK1/2 and p38α MAPK [21] in human chondrocyte cultures. For this purpose, we employed C28/I2 immortalized chondrocytes, which has been reported to retain a stable chondrogenic phenotype [22,23] as well as normal human chondrocytes to quantify the MAPK signaling cascade endpoints of unphosphorylated ERK1/2, JNK1/2, and p38α MAPK and phosphorylated ERK1/2, JNK1/2 and p38α MAPK. These were the principal SAPK/MAPK previously identified as activated by TNF-α and IL-1β in human osteoarthritis chondrocytes [21,24]. Thus, the overarching hypothesis that was tested in this study was that treatment of C28/I2 chondrocytes with rhIL-6, rhOSM or rhAPN would result in the phosphorylation of ERK1/2, JNK1/2 and p38α MAPK, without altering the level of total unphosphorylated ERK1/2, JNK1/2, and p38α MAPK.
The immortalized juvenile human chondrocyte line, C28/I2, was obtained from Dr. Mary B. Goldring (Hospital for Special Surgery/Weill Medical College of Cornell University, New York, NY). Normal human chondrocytes were purchased from Cell Applications, Inc. MAPK-specific antibodies reactive with U-ERK1/2, p-ERK/1/2 (phosphorylation sites, ERK1, Thr202/Tyr204, ERK2, Thr185/Tyr187); p38α kinase, P-p38α kinase (phosphorylation sites, Thr180/Tyr182); U-JNK1/2 and P-JNK1/2 (phosphorylation sites, Thr183/Tyr185) were purchased from Cell Signaling or R&D Systems. The β-actin antibody was from Santa Cruz Biotechnology. All secondary antibodies used for western blotting were purchased from Santa Cruz Biotechnology. The anti-U-STAT-antibodies were generally goat-anti-mouse antibodies, whereas the anti-P-STAT antibodies were generally goat-anti-rabbit antibodies.
Recombinant human (rh) IL-6 was from R&D Systems and recombinant human OSM and APN from ProSpecBio. The small molecule inhibitor of MEK1/2, 1,4-diamino-2,3-dicyano-1,4,bis[2-aminophenylthio]butadiene (U0126), was from Cell Signaling.
Confluent C28/I2 chondrocytes were incubated in DMEM/F12 medium (1:1) containing 0.1% fetal bovine serum (FBS) or 0.1%-FBS-containing DMEM/F12 and rhIL-6, rhAPN or rhOSM, (each at 50ng/ml) for 30 min. Normal human chondrocytes were incubated in DMEM/F12 (1:1) containing 0.1% FBS or 0.1%-FBS-containing DMEM/F12 and rhIL-6 (50ng/ml) for up to 60 min. U0126 was employed at 1μM, 10μM or 20μM to determine the extent to which upstream MEK1/2 activity was required for activating ERK1/2 in these cells as well as to test the specificity of U0126 by assessing the effect of U0126 on P-p38α MAPK and P-JNK1/2 as well. A set of control cultures containing dimethylsulfoxide (DMSO) was also included in this analysis because DMSO was used to fully dissolve U0126. Thus, U0126-containing cultures had an identical concentration of DMSO as did its control partner (i.e. without U0126).
Protein lysates were prepared by sonication of the cell pellets in ice-cold PBS and proteins separated by electrophoresis on SDS-10% polyacrylamide gels. ERK1/2, JNK, p38α MAPK, and β-actin were localized on western blots by probing with anti-unphosphorylated-specific (U) or anti-phosphorylated-specific (P) JNK, p38α or ERK1/2 antibodies along with an anti-β-actin antibody. We employed a sequential antibody-probing/stripping technique to measure either U-MAPK or P-MAPK antibody reactivity after which the immunoblot was stripped and reprobed with the β-actin antibody. The secondary antibodies had been applied in blocking buffer at the concentration recommended by the manufacturer. The immunoblots were developed with SuperSignal™ West Femto Maximum Sensitivity enhanced chemiluminescent (ECL) HRP substrate (Thermo Fisher Scientific). X-ray films were exposed for a comparable duration of time to the ECL-treated nitrocellulose membranes which were then subsequently developed in an X-ray film machine.
The migration rate, (Mr) of antibody-reactive protein bands was determined from the Mr of protein standards electrophoresed on the same acrylamide gel as the chondrocyte proteins. The ratio of U-ERK1/2, U-JNK1/2 or U-p38α MAPK and P-ERK1/2, P-JNK1/2 or P-p38α MAPK to β-actin was determined with the Metamorph® software program using the following strategy. The average intensity values from each western blot probed with the β-actin antibody were normalized to the highest value measured in control and treatment groups. The calculated values were then multiplied by the relative intensity values obtained for antibody-reactive bands in their affiliated blot (i.e. those blots probed with U-ERK1/2, U-JNK1/2, U-p38α MAPK, P-ERK1/2, P-JNK1/2 or P-p38α MAPK antibodies) in order to normalize each antibody-reactive band to its corresponding antibody-reactive band probed with β-actin. Finally, each of these calculated values were normalized to the experimental treatment (either lane 1 or lane 2 of each blot) to obtain the relative intensity value. Bar graphs were produced with Excel.
The difference in relative antibody-reactive band intensities between control and treated groups is expressed as +/-Δ%. Based on an initial inspection of data shown in the individual sets of bar graphs, the predicted difference between control and treated groups for biological responsiveness was set at Δ=+/-25%. The significance of these single point values were then analyzed by Chi-square (www.socscistatistics.com/tests/chisquare/default2.aspx) with a p-value of <0.05 considered as significant.
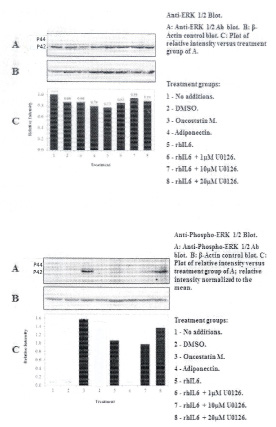
C28/I2 chondrocytes produced mainly U-ERK2 (i.e. p42ERK) as indicated on western blots by the reactivity of protein bands probed with the anti-U-ERK1/2 antibody (Figure 1 Top; Panel C). Constitutively activated p42ERK was not detected (Figure 1 Bottom; Lanes 1 and 2). It is also important to note that the p-ERK1/2 antibody employed in these western blots detects endogenous levels of p44/p42 ERK when p44ERK/p42ERK is phosphorylated either individually or dually at Thr202 and Tyr204 of p44ERK and Thr185 and Tyr187 of p42ERK. Thus, the overwhelming predominance for detecting p42ERK versus p44ERK (Figure 1, Bottom) in C28/I2 chondrocytes could reflect the antibody specificity for U-p44ERK versus U-p42ERK as well as the phosphorylation sites detected by the P-ERK1/2 antibody as suggested by the heavily skewed ratio of U-p42ERK to U-p44ERK in favor of U-p42ERK.
The most significant effect on activation of p42ERK was found after treating C28/I2 chondrocytes with rhOSM (Figure 1, Bottom Panel) (Table 1) whereas total U-p42ERK was unchanged (Figure 1, Top Panel). Treating C28/I2 chondrocytes with rhIL-6, but not rhAPN also increased P-p42ERK (Figure 1, Bottom Panel). Of note, incubation of C28/I2 chondrocytes with rhIL-6 plus U0126 (1μM) caused a significant reduction in P-p42ERK (χ2=29.3) which was not found at higher concentrations of U0126. Importantly, rhIL-6 plus U0126 did not change total U-p42ERK (Figure 1, Top Panel) (Table 1).
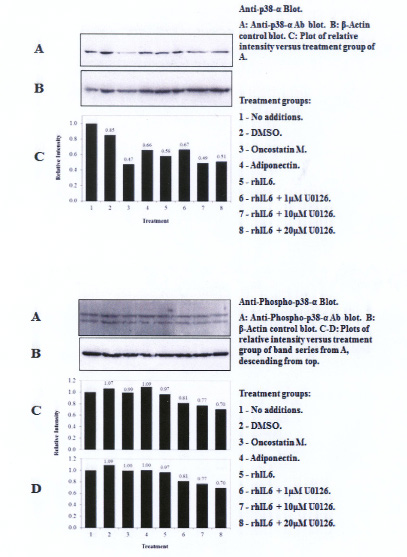
A single U-p38α MAPK antibody-reactive band migrating at ~38kDa was detected on western blots with the U-p38α MAPK antibody (Figure 2). However, constitutively activated p38α MAPK was detected at 0 min with the anti-P-p38α antibody (Figure 2A, Bottom Panel, lanes 1 and 2). These P-p38α MAPK reactive bands migrated to ~55kDa (i.e. P-p38α55kDa MAPK and P-p38α38kDa MAPK). Of note, incubation of C28/I2 chondrocytes with rhIL-6, rhOSM or rhAPN significantly reduced the content of the U-p38α reactive band. However, rhIL-6, rhOSM or rhAPN had no effect on either the P-p55kDa or the P-p38kDa MAPK reactive bands. Treating C28/I2 chondrocytes with rhIL-6 plus U0126 (20μM) reduced the content of total U-p38α MAPK, P-p38α55kDa MAPK and P- p38αp38kDa (Table 2). However, this effect was not detected at lower concentrations of U0126 (Table 2).
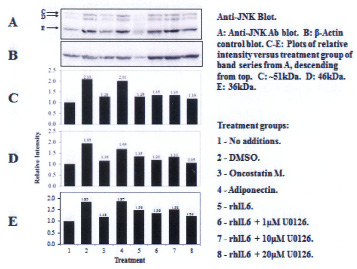
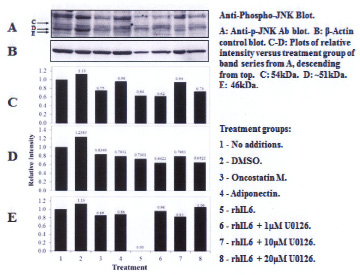
Three proteins migrating at Mr, ~51kDa, ~46kDa and ~36 kDa were reactive with the U-JNK1/2 antibody (Figure 3). Constitutive P-JNK was detected at 0 min (Figure 4, lanes 1 and 2). Treatment of C28/I2 chondrocytes with rhAPN resulted in a significant increase in total U-JNK51kDa and U-JNK46kDa. By contrast, treating C28/I2 chondrocytes with rhIL-6 or rhAPN significantly increased only P-JNK51kDa. Conversely, incubation with rhOSM had no effect on P-JNK51kDa. Of note, C28/I2 chondrocytes incubated with rhIL-6 plus U0126 significantly reduced U-JNK51kDa and U-JNK46kDa with variable effects on P-JNK51kDa and P-JNK46kDa (Table 3). However, only treatment with rhAPN significantly increased U-JNK36kDa (Figure 3) with no effect on P-JNK46kDa while rhIL-6 significantly increased P-JNK46kDa. The combination of rhIL-6 plus U0126 significantly decreased P-JNK46kDa which was not concentration-dependent (Table 4). U-JNK1/2 was also detected on western blots of proteins from normal human chondrocyte probed with the anti-U-JNK1/2 antibody where only the ~51kDa and ~46kDa JNK forms were found (Figure 5). After 30 min, rhIL-6 significantly decreased U-JNK51kDa but not U-JNK46kDa (Table 5). Constitutive P-JNK51kDa and P-JNK46kDa (Table 6) were detected at 0 time (Figure 6; lanes 1 and 2), however treatment of C28/I2 chondrocytes with rhIL-6 for various time periods increased P-JNK47kDa but not P-JNK51kDa (Figure 6 and Table 5).
The C28/I2 cell line of immortalized juvenile human chondrocytes produced primarily p42ERK as well as several forms of JNK1/2 and p38α MAPK which were detected on western blots with MAPK-specific antibodies. In that regard, the JNK1/2 and p38α MAPK forms differed in Mr from the predominant JNK1/2 form previously shown to be synthesized by human chondrocytes isolated from osteoarthritic cartilage [24-26]. One of these JNK forms migrated to a position consistent with 36kDa suggesting that JNK36kDa could be an alternative form of JNK produced by the C28/I2 human immortalized chondrocytes. In that regard, Zing, et al. [27] showed that 2 major isoforms of chicken JNK, namely JNK1 and JNK2 to which were assigned Mr's of 46kDa and 36 kDa, respectively, were found to be produced by chicken macrophages infected with the H9N2 virus. Furthermore, Zing, et al. [27] concluded that these JNK forms were consistent with several of the mammalian JNK isoforms reported by other groups. It should also be noted that some of the immortalized human chondrocyte lines were originally produced following transfection of primary costal cartilage chondrocyte cultures with the large T antigen of Simian virus 49 which yielded the T/C28a2 chondrocyte line from which C28/I2 was eventually produced [22].
In contrast to JNK, C28/I2 chondrocytes produced the ~38kDa form of U-p38α MAPK which was identical in size to the p38 MAPK previously reported to be produced by osteoarthritis human chondrocytes [24]. Importantly, C28/I2 chondrocytes produced constitutively activated P-p38α MAPK and P-JNK1/2, but not P-p42ERK.
The finding of constitutively phosphorylated p38α MAPK, JNK51kDa and JNK46kDa in the C28/I2 human chondrocyte line may merely be characteristic of the continuously proliferating phenotypic signature of this cell line. However, the absence of constitutive P-p42ERK distinguished p42ERK activation from p38α MAPK and JNK1/2 activation in this human chondrocyte line, perhaps making ERK1/2 a more appropriate choice for measuring the SAPK/MAPK response to pro-inflammatory cytokines and growth factors in this human chondrocyte cell line in vitro. Just as important was the novel finding that the JNK1/2 response to rhIL-6 by C28/I2 chondrocytes was similar to that found for normal human chondrocytes.
The finding that rhAPN did not significantly activate p42ERK also represents a novel finding regarding the interaction between APN and human chondrocytes. Previously published results with other cell types including human hepatocytes [28], MIN pancreatic β-cells [29] and neutrophils [30] as well as adipose tissue [31] had shown that APN activated ERK1/2. However, a careful analysis of the PubMed database failed to reveal any published papers indicating that treatment of human chondrocytes with rhAPN resulted in ERK1/2 phosphorylation.
Additional studies will be required to determine the extent to which activation of P42ERK by rhIL-6 or rhOSM is associated with regulating apoptosis, and/or AP-1 activity, the latter having implications for MMP gene expression. If these events are regulated by IL-6 or OSM they would be highly relevant to cartilage damage as a consequence of RA progression. However, it remains to be determined what the significance of the finding that rhIL-6, rhOSM reduced the ratio of U-p38α to β-actin or for that matter what the structural relationship is between the P-p38α55kDa band and P-p38α MAPK. It also should be noted that Aida, et al. [32] had previously shown that IL-6 and soluble IL-6 receptor stimulated normal human chondrocyte P-STAT3 and P-ERK. Stimulation of P-STAT3 and P-ERK was associated with increased MMP-1, MMP-13, tissue inhibitor of metalloproteinases-1 and plasminogen activator inhibitor-l gene expression.
The MEK1/2 inhibitor, U0126 did not alter the ratio of U-p42ERK to β-actin. However, U0126 at a concentration of 1μM did reduce the ratio of P-p42ERK to β-actin with less of an effect of U0126 measured at higher concentrations. This result would not have been expected if the effect of U1026 on P-ERK1/2 was totally U0126-concentration-dependent. Thus, these results may be due, in part, to how U0126 behaves when incubated with these immortalized human chondrocytes, including the unanticipated finding that in some cases, U0126 reduced the ratio of U-p38α MAPK and U-JNK1/2 to β-actin. In that regard, the finding of visible microscopic evidence of C28/I2 chondrocyte "blebbing" in rhIL-6 plus U0126-treated cultures at concentrations of 10μM and 20μM should be taken into account when interpreting these overall results with U0126. Therefore, additional studies of potential U0126-induced C28/I2 chondrocyte "cytotoxic" effects at higher U0126 concentrations will have to be conducted. Pertinent to this contention was finding that rhTNF-α-induced apoptosis in C28/I2 chondrocytes, as measured by the TUNEL assay, was further increased when C28/I2 chondrocytes were incubated with rhTNF-α plus U0126 at 10μM (unpublished data). It was also important to note that U0126 in combination with rhIL-6 for the most part also inhibited phosphorylation of the various forms of P-p38α MAPK and P-JNK1/2 produced by C28/I2 chondrocytes (Tables 2, 3 and 4). This result suggested the possibility that MEK1/2 activity could be involved in regulating the activation of p38αMAPK and JNK1/2 as well as its more defined activity as an inhibitor of ERK1/2 phosphorylation. Alternatively, these results may also mean that U0126 at 10μM and 20μM exhibited non-specific MEK/MAPK inhibitory activity.
Finally, we showed that rhIL-6 increased the ratio of U-JNK51kDa and U-JNK46kDa to β-actin in C28/I2 chondrocytes which should also warrant further study. We also need to clarify the structural relationship between the U-JNK36kDa form (which was reactive with the U-JNK1/2 antibody) and the P-JNK51kDa and P-JNK46kDa forms of JNK. It may also be relevant to an understanding of changes that occur in chondrocyte-related activity relative to RA progression to recognize the significance of the increase in U-JNK1/2 in response to rhIL-6, rhOSM and rhAPN. Thus, U-JNK1/2 may possess biological activity for regulating human chondrocyte gene expression as was previously reported for cytokine-inducible unphosphorylated-STAT proteins [33]. Importantly, the decrease in the U-JNK51kDa to β-actin ratio after treatment of normal human chondrocytes with rhIL-6 (Table 5) was dissimilar to that found for C28/I2 chondrocytes. This is likely to indicate differing responses to rhIL-6 between immortalized human chondrocyes and normal human chondrocytes.
Immortalized cell lines of human chondrocytes, exemplified herein by the C28/I2 chondrocyte line, have a definite utility in articular cartilage research because these cells can be employed for determining chondrocyte responses to cytokines and growth factors relevant to inflammation, RA and osteoarthritis In addition, these immortalized chondrocyte lines are easier to maintain as continuous cultures in the laboratory setting compared to primary human chondrocyte cultures which are, for the most part, obtained from individually discarded surgical specimens from patients who may have received various antecedent drug regimens which could irreversibly alter chondrocyte metabolism.
However, the results of one aspect of this study provided the initial evidence that the JNK1/2 response to rhIL-6 in normal human chondrocytes differed from C28/I2 chondrocytes. Therefore, this result alone should serve as an appropriate caveat for employing C28/I2 chondrocytes as a surrogate for normal human chondrocytes with respect to their use for translational research going forward.
The western blot results also revealed that C28/I2 chondrocytes produced constitutively phosphorylated p38α MAPK and JNK1/2. Thus, this finding differed from those previously reported for human chondrocytes isolated from surgical specimens of osteoarthritic knee cartilage where no constitutively activated p38α MAPK or JNK1/2 was detected by western blotting. However, in contrast to p38α MAPK and JNK1/2, phosphorylation of ERK1/2 was found only after rhIL6 or rhOSM treatment of C28/I2 chondrocytes, making this human chondrocyte line potentially useful for examining how ERK1/2 activation alters chondrocyte gene expression in response to these cytokines.
Overall, the changes seen in the level of total U-JNK1/2 and U-p38α MAPK in C28/I2 chondrocytes in response to rhIL-6, rhOSM and rhAPN also suggested that these cells could be employed to determine if U-JNK1/2 and U-p38α MAPK play any role in regulating human chondrocyte gene activation.
This study was supported by a contract (#ACT077) to Charles J. Malemud, Ph.D. and CWRU from Genentech/Roche Group and by the CWRU Visual Sciences Core Center Grant (National Eye Institute; P30 EY11373).

![]()

 |
| Figure 1: Western blot analysis: Effect of rhIL-6, rhIL-6 + U0126, rhOSM or rhAPN on U-ERK1/2 and P-ERK1/2 in C28/I2 chondrocytes |
 |
| Figure 2: Western blot analysis: Effect of rhIL-6, rhIL-6 + U0126, rhOSM or rhAPN on U-p38α MAPK and P- p38α MAPK in C28/I2 chondrocytes |
 |
| Figure 3: Western blot analysis: Effect of rhIL-6, rhIL-6 +U0126, rhOSM or rhAPN on U-JNK1/2 in C28/I2 chondrocytes |
 |
| Figure 4: Western blot analysis: Effect of rhIL-6, rhIL-6 +U0126, rhOSM or rhAPN on P-JNK1/2 in C28/I2 chondrocytes |
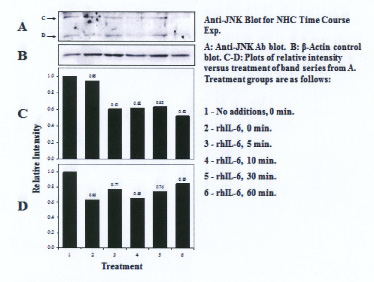 |
| Figure 5: Western blot analysis: Effect of rhIL-6, rhIL-6 +U0126, on U-JNK1/2 in normal human chondrocytes (NHC) |
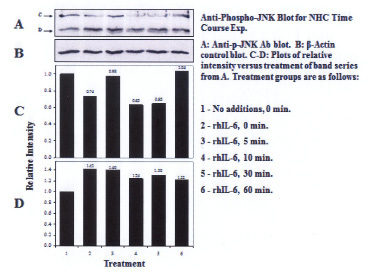 |
| Figure 6: Western blot analysis: Effect of rhIL-6, rhIL-6 +U0126, on P-JNK1/2 in normal human chondrocytes (NHC) |
| Cytokine | U-ERK2 | P-ERK2 | χ2 U-ERK25 | χ2 P-ERK25 |
| rhIL-6 | -29.91 | +1071 | 0.36 | 32.7* |
| rhIL-6 + U01262 | -1.11 | -1001 | 19.8* | 29.3* |
| rhIL-6 + U01263 | +9.31 | -9.21 | 6.5* | 6.5* |
| rhIL-6 + U01264 | +2.31 | +28.91 | 17.4* | 0.23 |
| rhOSM | -16.31 | +1571 | 1.64 | 56.9* |
| rhAPN | -26.61 | 01 | 0.06 | 1.31 |
| 1 Δ% 2 1μM 3 10μM 4 20μM 5 χ2 analysis based on predicted Δ% = ±25 *χ2 value for p<0.05 is 3.841 Table 1: IL-6-type Cytokines Primarily Activate p42ERK (ERK2) in C28/I2 Chondrocytes |
||||
| Cytokine | U-p38α 38KDa | P-p38α 55KDa | P-p38α 38KDa | χ2 U-p38α5 | χ2 P-p38α5 55KDa | χ2 P-p38α5 38KDa |
| rhIL-6 | -72.41 | -3.11 | -3.11 | 13.21* | 0.995 | 0.995 |
| rhIL-6 + U01262 | -26.91 | -32.11 | -34.61 | 0.061 | 0.67 | 1.28 |
| rhIL-6 + U01263 | -73.51 | -39.01 | -41.61 | 16.70* | 2.33 | 3.24 |
| rhIL-6 + U01264 | -66.71 | -52.91 | -55.71 | 13.40* | 7.30* | 8.55* |
| rhOSM | -112.81 | -1.01 | 01 | 19.89* | 1.20 | 1.31 |
| rhAPN | -51.51 | +9.01 | 01 | 9.54* | 0.505 | 1.31 |
| 1 Δ% 2 1μM; 3 10μM; 4 20μM 5 χ2 analysis based on a predicted Δ% = ±25 *χ2 value for p<0.05 is 3.841 Table 2: Effect of IL-6-type Cytokines on U-p38α MAPK and P-p38α MAPK |
||||||
| Cytokine | U-JNK1 | U-JNK2 | P-JNK1 | P-JNK2 | χ2 U-JNK15 | χ2 U-JNK25 | χ2 P-JNK15 | χ2P-JNK25 |
| rhIL-6 | +28.01 | +35.01 | -56.31 | -56.31 | 0.13 | 1.3 | 8.5* | 1.5 |
| rhIL-6 + U01262 | -62.21 | -54.21 | -.91.91 | -.91.91 | 11.20* | 6.90* | 25.30* | 24.40* |
| rhIL-6 + U01263 | -55.91 | -46.61 | -27.01 | -27.01 | 8.53* | 4.98* | 0.061 | 7.30* |
| rhIL-6 + U01264 | -75.61 | -85.71 | -50.61 | -50.61 | 17.61* | 22.42* | 6.50* | 22.90* |
| rhOSM | +28.01 | +38.01 | -33.31 | -33.31 | 0.134 | 2.05 | 0.856 | 0.453 |
| rhAPN | +1041 | +69.01 | -4.21 | -4.21 | 31.21* | 14.33* | 13.40* | 0.012 |
| 1 Δ% 2 1μM 3 10μM 4 20μM 5 χ2 analysis based on a predicted Δ%=±25 *χ2 value for p<0.05 is 3.841 Table 3: Effect of IL-6-type Cytokines on U-JNK1 (P46)/U-JNK2 (P51) and P-JNK1 (P46)/P-JNK2 (P51) |
||||||||
| Cytokine | U-JNK36kDa5 | P-JNK36kDa5 | χ2 U-JNK36kDa | χ2 P-JNK36kDa |
| rhIL-6 | +20.01 | 01 | 0.45 | 22.5* |
| rhIL-6 + U01262 | -40.81 | -55.61 | 2.93 | 8.6* |
| rhIL-6 + U01263 | -38.71 | -127.71 | 2.33 | 42.9* |
| rhIL-6 + U01264 | -47.51 | -72.61 | 5.35* | 16.2* |
| rhOSM | +13.01 | -19.01 | 3.19 | 0.7 |
| rhAPN | +87.01 | -13.61 | 22.90* | 2.6 |
| 1 Δ% 2 1μM 3 10μM 4 20μM 5 χ2 analysis based on predicted Δ% = ±25 *χ2 value for p<0.05 is 3.841 Table 4: Effect of IL-6-type Cytokines on the 36kDa Form of U-JNK (U-JNK36kDa) and 36kDa Form of P-JNK (P-JNK36kDa) |
||||
| Time (min) | U-JNK2-51kDa | U-JNK1-46kDa | χ2 U-JNK22 | χ2 U-JNK12 |
| 0 | -5.31 | -63.91 | 11.7* | 12.0* |
| 5 | -63.91 | -29.91 | 11.6* | 0.4 |
| 10 | -61.31 | -53.81 | 10.7* | 7.7* |
| 30** | -61.31** | -35.11** | 10.7* | 1.2 |
| 60 | 92.31 | -17.61 | 25.3* | 0.9 |
| Table 5: Effect of rhIL-6 on U-JNK1 (46kDa) and U-JNK2 (51kDa) in Normal Human Chondrocytes | ||||
| Time (min) | P-JNK1-51kDa | P-JNK2-46kDa | χ2 P-JNK22 | χ2 P-JNK12 |
| 0 | -35.11 | +42.01 | 1.2 | 3.2 |
| 5 | -7.51 | +40.01 | 7.6* | 2.6 |
| 10 | -61.31 | +34.01 | 10.7* | 1.1 |
| 30** | -53.81** | +30.01** | 7.7* | 0.4 |
| 60 | +3.01 | +22.01 | 15.3* | 0.2 |
| 1 Δ% 2 χ2 analysis based on predicted Δ% = ±25 *χ2 value for p<0.05 is 3.841 ** 30 min was the incubation time for measuring the C28/I2 chondrocyte response to rhIL-6 Table 6: Effect of rhIL-6 on P-JNK1 (51kDa) and P-JNK2 (46kDa) in Normal Human Chondrocytes |
||||




































