Open Access
Research Article
Max Screen >>
ISSN: 2454-499X
Copyright: © 2022 Nankya I. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Objective: To determine the prevalence of minority drug resistance variants in the protease and Gag regions among patients failing a protease inhibitor (PI) based regimen with or without a susceptible genotype based on Sanger sequencing technology.
Methods: Samples were obtained from patients who were failing on a protease inhibitor-based regimen (n = 500). Sanger based sequencing was performed as part of the standard of care. Mutation analysis was performed using the Stanford HIV drug Resistance database. A subset of these patient samples was grouped into two categories: those failing a PI based with mutations in the protease region (n = 100) and those failing on a PI based regimen without mutations in the protease region (n = 128). These samples were then analyzed in the protease and Gag regions using Next Generation Sequencing (NGS) technology and analysis of the drug resistance mutations was performed at the 20% and 1% cutoffs.
Results: An initial analysis of the protease region for patients failing with drug resistance mutations revealed that most patients harbored mutations that confer resistance to Lopinavir and Atazanavir, but these mutations had little effect on Darunavir. Furthermore, NGS revealed that in patients failing with and without drug resistance mutations, minority drug resistance mutations were present at each of the drug resistance codons and at codons that confer multi-drug resistance to protease inhibitors. Further analysis of the Gag gene revealed more genetic diversity among patients failing with no mutations in the protease as evidenced by the proportion of polymorphisms at each codon.
Conclusion: Based on Sanger sequencing, a proportion of patients fail a PI based regimen with a susceptible genotype. However, these patients harbor minority variants in the protease and numerous polymorphisms in the Gag region which when combined these could explain their poor response to therapy. Therefore, in order to improve patient care in low resource settings, there is need to adapt NGS as the standard genotyping technique so that minority variants are captured much earlier. In addition, since mutations in the Gag region also play a role in response to PIs, this region should be included in the routine monitoring for response to therapy in patients on a PI based regimen.
Keywords: Next Generation Sequencing, Minority Variants, Gag Polymorphisms
In order for ART to effectively control viral replication, it is imperative that it is taken consistently as prescribed [1]. The challenge however is that due to a number of reasons, insufficient drug levels are delivered into the system [2]. This then means that the available drug levels are not sufficient to control viral replication [3]. Therefore, viral strains which are able to replicate in the presence of drug pressure emerge [3]. These variants are the drug resistant strains. The population of these variants slowly builds up over time from minority variants which cannot be detected by the conventional Sanger sequencing assays to predominant viral populations which have the potential of causing multi-drug resistance [4].
The extensive use of ART and multiple combination treatments has favored the emergence of novel patterns of mutations conferring drug resistance. Although antiretroviral drugs are designed based on the subtype B variants, they work effectively against all the other known subtypes [5]. However, with more than 30 million patients infected with non-subtype B HIV-1 receiving HAART, the need for more non-subtype B specific studies cannot be over emphasized since studies have shown some subtype specific differences with regard to certain mutation profiles [6-11].
Protease inhibitors have the advantage of a high genetic barrier therefore if taken as recommended, resistance to PIs emerges after prolonged periods of use [12]. Protease inhibitors bind to the various polypeptide cleavage sites within the Gag and Gag-Pol polyproteins and so prevent the protease enzyme from cleaving these proteins into individual functional mature proteins. In this way the released viral particles are immature and noninfectious [13-15]. They act by binding to the various cleavage sites within the Gag and Gag-Pol polyproteins to prevent proteolytic cleavage of these non-functional proteins into functional mature proteins during or shortly after release from the cell [13-15]. In order to obtain these functional proteins, the protease cleaves the Pr55gag at: MA/CA, CA/p2, p2/NC, NC/p1, and p1/p6 [16]. With this huge number of cleavage sites, substrates, intermediates and products, it is possible that mutations occurring elsewhere in any of these regions could confer drug resistance to PIs without necessarily showing mutations in the protease region [16-26]. Studies have shown that mutations in the Gag cleavage sites are associated with resistance to PIs [22-28]. However, the effect of mutations in other parts of the Gag and Gag-Pol have not been fully studied. With such a complex cascade of reactions in the Gag and Gag-Pol involving the protease, it likely that many mutations in there may affect response to protease inhibitors.
In this study, using Next Generation Sequencing (NGS) technology on a cohort that is comprised mainly of subtype A and D patients failing on a protease inhibitor (PI) based regimen with or without major drug resistance mutations in the protease region, we analyzed for the presence of minority variants in both the protease and Gag regions.
The Joint Clinical Research Centre (JCRC) is an HIV treatment, care and research center that has been offering antiretroviral therapy to HIV positive patients for the past thirty years. In the early 1990s, although treatment was offered, it was suboptimal, mainly mono and dual therapy. Triple therapy was introduced in the late 90s but still not readily available to the general population. With the global roll out of ART in 2004, treatment became universally available to all patients with the first line regimen comprising of two NRTIs plus an NNRTI. The PI based second line regimen mainly consisted of 2 NRTIs and a boosted PI. Patients were able to have full access to virological monitoring in order to determine response to therapy and this included a viral load test as well as an HIV drug resistance test for those with a viral load greater than 1000 copies/ml. Ethical clearance for this analysis was provided by the institutional review board (protocol, EM-10-07) at JCRC, Kampala Uganda.
Using the JCRC patient care database, all patients failing on a PI based regimen as evidenced by a viral load greater than 1000 copies/ml were selected. For a sample to be included in the analysis, it had to have an HIV genotype result in the protease-reverse transcriptase region based on Sanger sequencing technology. From among these patient results, two groups of samples were obtained: 1). Patients failing on a PI based regimen with evidence of drug resistance mutations in the protease region (n = 100, 2). Patients failing on a PI based regimen without evidence of drug resistance mutations in the protease region n = (128). Anonymized patient databases containing patient demographics and laboratory results were merged with HIV-1 drug resistance databases after stripping samples of identifiers in accordance with the IRB approval.
Plasma samples from patients were obtained as part of the routine monitoring for response to ART. RNA was extracted from plasma samples obtained from the patient categories mentioned using the Qiagen viral RNA extraction kit according to the manufacturer’s instruction (Qiagen Inc. Chatsworth, California, USA). The Gag region was amplified using conserved primers to obtain a 1200 base pair fragment.
PCR products encompassing the Gag coding region of the HIV genome were sequenced using NGS as described by Gibson et al. Briefly, amplicons were purified (Agencourt AMPure XP, Beckman Coulter) and quantified (Bioanalyzer DNA 7500, Agilent Technologies) prior to using the Ion Xpress Fragment Library Kit (Life Technologies, Carlsbad CA) to construct a multiplexed library for shotgun sequencing on the Ion Personal Genome Machine (PGM, Life Technologies). For that, 33ng of purified DNA amplicons was randomly fragmented and blunt-ends repaired using the Ion Shear Plus Reagent (Life Technologies) followed by DNA purification (Agencourt AMPure XP, Beckman Coulter). The P1 adapter and one of 12 barcodes were ligated to the repaired fragment ends prior to DNA purification (Agencourt AMPure XP, Beckman Coulter). DNA fragments were then selected by size (i.e., 280 to 320 bp, Pippin Prep, Life Technologies) and each barcoded library, was purified (Agencourt AMPure XP, Beckman Coulter) and normalized using the Ion Library Equalizer Kit (Life Technologies). All thirteen barcoded DNA libraries, corresponding to the patient-derived amplicons plus the HIV- 1NL4-3 control, were pooled in equimolar concentrations and templates prepared and enriched for sequencing on the Ion Sphere Particles (ISPs) using the Ion OneTouch 200 Template Kit v2 (Life Technologies) in the Ion OneTouch 2 System (Life Technologies). Templated ISPs were quantified (Qubit 2.0, Life Technologies) and loaded into an Ion 316 Chip (Life Technologies) to be sequenced on the Ion PGM using the Ion PGM Sequencing 200 Kit v2 (Life Technologies). Following a 4 hour and 20 minutes sequencing run, signal processing and base calling was performed with Torrent Analysis Suite version 3.4.2.
Read mapping and variant calling: Reads were mapped and aligned against sample-specific reference sequences constructed for the gag (gag-p2/NCp7/p1/p6) using the DEEPGEN Software Tool Suite as described. The frequency of each amino acid present in each HIV-1 genomic position was calculated and summarized in a graphical interface with particular focus on sites of known drug resistance based on the latest edition of the IAS-USA HIV Drug Resistance Mutations list [57]. A list of the amino acids at these positions, and their frequencies, was exported as a tabulated text file and used with the HIVdb Program Genotypic Resistance Interpretation Algorithm from the Stanford University HIV Drug Resistance Database (https://hivdb.stanford.edu) to infer the levels of susceptibility to protease, reverse transcriptase, and integrase inhibitors.
Subtype analysis was performed for the Gag and protease regions. For both the Gag and protease regions, subtype A was slightly more predominant than subtype D (Figure 1).
For patients failing on a PI based regimen with major mutations in the protease region, we compared the level of drug resistance (that is: susceptible, low-level resistance and intermediate to high level resistance) for the commonly used boosted PIs namely Lopinavir, Atazanavir and Darunavir. About 80% of these patients exhibited intermediate to high level resistance to Lopinavir and Atazanavir with only about 10% still susceptible to these to drugs (figure 2A). On the other hand, about 50% of these patients harbored virus that was still susceptible to Darunavir with about 30% of these having intermediate to high level resistance to the same drug (figure 2A). When individual mutations at each codon for each of these drugs were analyzed, we found that about 45% of patients harbored major mutations that confer resistance to Lopinavir and Atazanavir. For each of these two drugs, mutations such as M46, I54 and I82 which confer multi-drug resistance (MDR) to all the other PIs were present at cumulative proportions of about 40% (figure 2B). On the other hand, 10% of the patients harbored major mutations that conferred resistance to Darunavir. For Darunavir, mutations such as I31, M46, L76 and I84 were present in about 2% of the patients. Although these are major PI mutations, they do not confer high level of resistance to Darunavir and are not associated with multi-drug resistance (figure 2B).
Next, using NGS, we looked for presence of minority variants below the 20% Sanger cut off that confer drug resistance to PIs among patients harboring major PI mutations. At each of the codons, minority variants were detected at varying proportions implying that although these patients exhibited detectable major drug resistance mutations above the 20% cut off, still among the same patients there were minority mutations present at proportions lower than the Sanger limit of detection (figure 3A). Using the same approach, we analyzed samples from patients failing on a PI based regimen with a susceptible genotype. More evident than among the patients failing with major PI mutations was the fact that at each of the drug resistance positions (including positions that confer MDR to PIs), at each position, there was on average 10% of the patients’ exhibiting mutations below the 20% cut off (figure 3B).
Various studies have documented the role of mutations in the Gag region in patients failing a PI based regimen. Most of the mutations have been found at the MA/CA, CA/p2, p2/NC, NC/p1, and p1/p6 cleavage sites. We analyzed the matrix, capsid, p2, p7(NC), and p1 using NGS. For each of these regions, we compared genome stability between patients failing with mutations and those failing without any mutations in the PI region (Figure 4).
For the Matrix, region of the Gag, comparison of the patients failing with no mutations to those failing with mutations revealed that at almost every codon, the proportion of patients exhibiting mutations was much higher in patients failing with no mutations in the protease region compared to those failing with mutations in protease (figure 4A). The same results were seen in the capsid (figure 4B). Interestingly, the p1 region gave the most significant findings where almost all positions apart from one the mutation proportions were higher in patients failing with no mutations in the PI (figure 4D). The NC (p7) region however did not show that much difference between those failing with mutations and those without mutations in the protease region (figure 4C).
Despite the high genetic barrier of PIs, mutations first develop in the active site of the protease followed by a step-wise accumulation of mutations surrounding the active site, then at cleavage sites and at non-cleavage sites of the Gag-pol polyprotein which compensate for replication defects and increase phenotypic resistance [14, 29, 30]. Therefore, there is increasing evidence of the role played by mutations outside of the protease and the possibility of still undefined mechanisms of resistance to PIs [16, 17, 19-25, 27, 31].
In this study we have shown that among the new generation PIs under current use, a high proportion of patients failing on a PI based regimen present with mutations that confer high level resistance to boosted lopinavir and atazanavir [32-34]. However, most of these patients are still susceptible to darunavir. As previously shown by Schnell et al, mutations that confer resistance to lopinavir also confer cross resistance to atazanavir in the same proportions but these mutations have little or no effect to the susceptibility of darunavir [34]. Most of these patients had mutations such as M46, I54 and I82 which confer high level resistance to these two drugs and multi-drug resistance to all the old generation PIs but these mutations do not affect darunavir. Three mutations were common to the three drugs: V32, I47, and I84. Aoki et al showed that although the V32I mutation is critical for darunavir resistance as it is associated with high level resistance, it is also associated with reduced viral fitness which means that this drug can still be used even in the presence of that mutation [29]. We found that the prevalence of this mutation was as low as 2% in each of the three drugs.
The high barrier to darunavir resistance has been demonstrated in numerous studies which have confirmed that the development of darunavir resistance associated mutations (RAMs) and phenotypic resistance was very rare [35-43]. Brown et al showed that the prevalence of darunavir RAMs among commercially tested samples remained low and stable between 2010 and 2017 and a high proportion of these exhibited phenotypic susceptibility to darunavir [44]. In fact, for this reason, darunavir is currently being used in patients who have failed on other PIs. Chaussade et al showed that among patients who were failing a PI based regimen with evidence of drug resistance mutations, only 20% of these developed new mutations when put on darunavir [45].
Analysis of PI RAMs at each codon using NGS for patients failing with a resistant genotype and those failing with a susceptible genotype revealed that in both these patient populations, drug resistance mutations were present at levels not detectable by the conventional Sanger sequencing technique. What this means is that even in patients exhibiting certain mutations that confer resistance, other RAMs are slowly evolving from minority variants to a level detectable by the conventional Sanger method. This finding was even more pronounced in patients failing without detectable RAMs based on Sanger. It is very evident that various proportions of these patients exhibit minority variants. These variants slowly evolve to become the majority population. It is also possible that even at levels below 20%, these mutations still have the ability to influence response to therapy hence the reason for patients failing to control viremia but in the absence of RAMs above the 20% cut off as determined by the conventional methods. This finding is supported by Kyeyune et al who showed that there is a proportion of patients who fail an RT containing regimen with a susceptible genotype based on Sanger sequencing technology but with resistance mutations below 20% using NGS [46]. This has been confirmed by Samuel et al who showed that the presence of minority variants that confer resistance to nevirapine is associated with treatment failure [47]. Hence, for RAMs to affect response to therapy, they do not have to be at a level greater than 20% of the viral population. Although various studies still doubt the significance of these minority variants [48], it is clear that they do play an important role in the treatment outcomes we see in a proportion of the patients receiving ART [46]. On the flip side, it is possible that the presence of these minority variants especially in patients failing with a susceptible genotype could be as a result of poor adherence. What poor adherence does is that in the presence of low drug levels, drug resistant variants emerge in the population, they appear as the evolving viral intermediates (EVI) [46] which then undergo evolution to become the dominant resistant variants. If poor adherence gets to the level where no drug pressure is being exerted, the mutant variants then slowly revert to wild type however, the EVI will still be detected at very low levels. These then may become fully dominant if full drug pressure is exerted [46]. Therefore, it is possible that the minority variants seen on NGS are either the beginning of the evolution of these variants or fading variants as the virus reverts to wild type. There is a possibility that both these theories apply in different patients where in some, the emerging low level viral mutations confer reduced response to therapy and in this case these variants slowly evolve to become the majority variants within the population. While in others, the fading mutations as the virus reverts to wild type in the absence of drug pressure in patients who have ceased taking ART account for the reduced response to therapy. In order to prove which of these two theories holds, longitudinal studies closely monitoring viral evolution using NGS need to be carried out. However, for ethical reasons, when such mutations reach 20% of the viral population, then a regimen switch is recommended.
In order to escape recognition by the PIs, HIV not only does it develop mutations in and around the protease active site but it also devises means of utilizing the Gag gene as an escape route by developing mutations at the various cleavage sites [17, 18, 21, 25, 27]. These mutations have been documented in causing drug resistance to PIs. In this study we show that in patients failing a PI based regimen the Gag gene is very dynamic and highly unstable as evidenced by the various mutations along the Gag region. We found that these mutations were more pronounced in patients failing without PIs RAMs further confirming that the Gag is under constant selection pressure at cleavage sites in patients with PI RAMs but also all through the Gag gene and more so in patients without drug resistance mutations in the protease region [31]. These findings suggest that other than the Gag cleavage site mutations, there are many more novel mutations that are likely to confer resistance to PIs within the different Gag regions. Site directed mutagenesis together with phenotypic assays need to be done in order to fully characterize these novel mutations. Following results from phenotypic assays, there may be need to include the Gag gene in routine testing for response to therapy by PIs since PI RAMs may not be sufficient to account for the poor response to therapy. Furthermore, based on our findings and those of others [46], NGS may be the best approach to determine response to therapy since we have seen that in patients failing with or without PI RAMs, minority variants are present and these may play an important role in explaining why some people fail on a regimen but with a susceptible genotype based on Sanger sequencing. Therefore, the presence of minority variants in the protease region together with the presence of a multitude of mutations at non-cleavage sites within the Gag region may explain why people fail a PI based regimen without evidence of RAMs in the protease region.
In summary, we show that minority variants present in patients failing on a PI based regimen with a susceptible genotype may contribute to treatment failure. These variants are present both in the protease and Gag regions of the genome. Theoretically these variants may be as a result of emerging or fading mutations but in each case, they contribute to treatment failure. The major challenge with these minority variant mutations is that they give a ‘false’ susceptible genotype based on Sanger sequencing which led physicians into recommending intensified adherence counseling (IAC). What then happens is that if these were fading mutations, with improved adherence, the minority mutations return to become the dominant variants. If the minority variants were emerging, improving adherence will increase the drug pressure and this will result in the minority variants ultimately becoming the dominant ones in a much shorter time. Therefore, the importance of NGS as the standard for patient care cannot be over emphasized as this will help pick these minority variants much earlier and help preserve the limited drug options.
Case Western Reserve University Centre for AIDS Research (CWRU/CFAR) through the CFAR Developmental Fund supported this research
This work was carried out in collaboration between all authors. Immaculate Nankya (IN) designed the study, performed data analysis and wrote the manuscript, Eva Nabulime (EN) performed all PCR and Sanger sequencing procedures, Fred Kyeyune performed all the NGS work, Miguel Quinones (MQ) did all the NGS data analysis and Cissy Kityo (CK) made edits and gave comments on the draft. All authors read and approved the final manuscript.

![]()

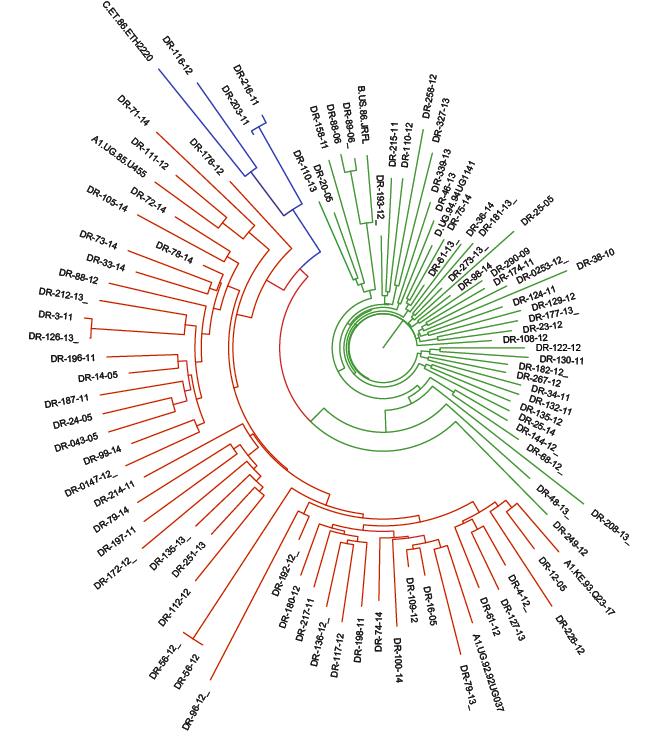 |
| Figure 1: Phylogenetic tree showing subtype distribution among patients failing a PI based regimen. Phylogenetic tree was constructed for patients failing on a PI based regimen by compiling sequences for the protease-reverse transcriptase (PR-RT) region and the Gag region. These sequences were obtained by Sanger sequencing. Sequences were aligned against known subtype references. Subtype distribution based on the PR-RT region (Fig 1A). Subtype distribution based on the Gag region (Fig 1B). |
 |
 |
| Figure 2: Response to therapy based on the three protease inhibitors under current use. Response to LPV, ATV and DRV was assessed. Level of resistance (susceptible, low-level resistance and intermediate to high level resistance) was assessed each of these three drugs using the Stanford HIV drug resistance database (Fig 2A). For each of the drugs, proportions of resistance occurring at each codon was assessed and a cumulative proportion of overall resistance for each drug was obtained (Fig 2B) |
 |
 |
| Figure 3: HIV drug resistance mutations occurrence at each codon of the protease gene. Using Next Generation Sequencing, samples from patients failing a PI based regimen were sequenced. Samples from patients failing a PI based regimen with mutations based on Sanger sequencing technology were sequenced in the protease to assay for minority variants that confer resistance to PIs. Minority (<20%) and majority (>20%) mutations at each codon were analyzed (Fig 3A). Samples from patients failing on a PI based regimen with a susceptible genotype based on Sanger technology were also sequenced by NGS and minority variants that confer resistance to PIs were analyzed by codon (Fig 3B). |
 |
 |
 |
 |
| Figure 4: Diversity within the Gag gene in patients failing a PI based regimen. Samples from patients failing a PI based regimen with mutations conferring resistance in the protease gene (in blue) and with susceptible genotype (in orange) based on Sanger technology were sequenced using NGS and analyzed at codon level for the different regions within the Gag. A comparison for the frequency of mutations was made between those harboring drug resistance mutations and those with susceptible genotype for the following regions: Matrix (Fig 4A), Capsid (Fig 4B), p7 (Fig 4C) and p1-sp2 (Fig 4D). |




































