Open Access
Review Article
Max Screen >>
ISSN: 2454-499X
Copyright: © 2016 Singh N. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
The currently available anti HIV agents have several drawbacks such as short half life, low bioavailability, poor CNS penetration and retention, hepatic first pass metabolism, undesirable side effects and frequent dosing regimen. Due to these drawbacks antiretroviral (ARV) drugs mostly oral formulations are inconvenience to the HIV patients and frequent administration of doses lead to poor patient compliance. So, these drawbacks give researchers tremendous opportunities to design and develop novel drug delivery systems to overcome the transport barriers and inherent elimination and metabolism problems associated with this anti HIV drugs. Novel drug delivery systems are developed by the application of the concepts and techniques of controlled release drug administration which not only extend the potent life of existing drug but also minimize the scope and expenditure of testing required for FDA approval and which make clinically already established drugs do their therapeutic best. The objective in the design of a controlled drug release system is to release a pharmacologically active agent in a predetermined, predictable and reproducible fashion. Many novel drug delivery systems have been developed e.g. Transdermal, Intrauterine, Intravaginal, Niosomes, Liposomes, Nanoparticles and Implants etc. These drug delivery systems have added a new dimension of optimizing the treatment of several disease conditions by modifying various pharmacokinetics parameters. These drug delivery system releases the drug either by zero order kinetics or by first order kinetics or by both simultaneously. The aim of this review is to provide a summary of the application of novel drug delivery systems to the delivery of anti-HIV drugs, specifically antiretrovirals. controlled/sustained and targeted/intracellular drug delivery are the important ones for antiretroviral (ARV) drugs which may useful for further research in future.
Keywords: AIDS; HIV; Antiretroviral therapy; Novel Drug Delivery Systems
AIDS is a collection of symptoms and infections resulting from the specific damage to the immune system caused by the human immunodeficiency virus (HIV) [1]. The late stage of the condition leaves individuals prone to opportunistic infections and tumors. Although treatments for AIDS and HIV exist to slow the virus’s progression, there is no known cure. HIV is transmitted through direct contact of a mucous membrane or the blood stream with a body fluid containing HIV, such as blood, semen, vaginal fluid, pre seminal fluid and breast milk [2]. Antiretroviral treatment reduces both the mortality and the morbidity of HIV infection, but routine access to antiretroviral medication is not available in all countries [3]. There is currently no vaccine or cure for HIV or AIDS. Therapy is based on avoiding exposure to the virus or an antiretroviral treatment directly after a highly significant exposure, called post-exposure prophylaxis (PEP) [4]. The major classes of antiretroviral agents are as follows:
• Nucleoside reverse transcriptase inhibitors (NRTI)
• Nucleotide reverse transcriptase inhibitors (nRTI)
• Protease inhibitors
• Non-nucleoside reverse transcriptase inhibitors (NNRTI)
• Integrase inhibitors
• Entry inhibitors (or fusion inhibitors)
• Maturation inhibitors
These are the first class of antiretrovirals. NRTIs are prodrugs that require intracellular activation to become NRTI-triphosphates. Once activated, the drug competes with endogenous deoxynucleotide-triphosphate and inhibits the activity of the reverse transcriptase, a viral enzyme involved in the synthesis of the viral DNA from its RNA [5]. All compounds of in this class are prodrugs which need to be converted intracellularly in the cytoplasm to their active form before exerting their antiviral activity. The active forms of these drugs are substrates for reverse transcriptase enzyme and they result in termination of DNA chain elongation of the retrovirus [4].
Tenofovir is the only FDA-approved nucleotide reverse transcriptase inhibitor. Since tenofovir is already monophosphorylated the drug skips the activation stage required by NRTI. No research works dealing with the relatively low bioavailability of the drug (25–30%) have been reported up to date [6].
They act at the end of the HIV life cycle to cause the formation of non- infectious immature virions. These agents represent a major advance in the management of HIV disease and have dramatically altered disease progression to AIDS [4].
They inactivate the HIV-1 reverse transcriptase enzyme by non-competitively binding directly to the HIV-1 reverse transcriptase structure likely at amino acid positions 100 and 103. These are not active against HIV-2. Unlike NRTIs these donot require activation by cellular phosphorylation [4].
They inhibit the enzyme integrase, which is responsible for integration of viral DNA into the DNA of the infected cell. There are several integrase inhibitors currently under clinical trial.
• Raltegravir
They interfere with binding, fusion and entry of HIV-1 to the host cell by blocking one of several targets.
• Maraviroc
• enfuvirtide
They inhibit the last step in gag processing in which the viral capsid polyprotein is the mature capsid protein. Because these viral particles have a defective core, cleaved, thereby blocking the conversion of the polyprotein into the virions released consist mainly of non-infectious particles. There are no drugs in this class currently available, though two are under investigation [7] (Table 1).
• Maraviroc
• enfuvirtide
The currently available ARV drugs associated with several disadvantages and inconveniences to the HIV patients. The delivery of drugs via oral route suffers from significant first-pass effect, variation of absorption and degradation in the gastrointestinal tract due to enzymes and extreme pH conditions leading to low and erratic bioavailability. Same time the duration of drug action also limited [13]. The metabolism/elimination and transport barriers will substantially reduce the effective amount of anti-HIV drug reaching the target action site. The half-life for several ARV drugs is short, which then requires frequent administration of doses leads to poor patient compliance [13].
Currently available anti HIV agents have following drawbacks:
1. Short half life
2. Low bioavailability
3. Poor CNS penetration and retention
4. Hepatic first pass metabolism
5. Undesirable side effects
6. Frequent dosing regimen
So, these drawbacks give researchers tremendous opportunities to design and develop novel drug delivery systems to overcome the transport barriers and inherent elimination and metabolism problems associated with these anti HIV drugs [4].
To avoid hepatic first-pass effect and intestinal degradation, various researches have made efforts on the delivery of drug. Delivery of nucleoside analogues through percutaneous absorption, rectal administration, oral buccal permeation, nasal absorption, intratracheal administration, using enteric-coated dosage form and co-administration with antacid are being studied. The novel drug delivery systems are developed by the application of the concepts and techniques of controlled release drug administration which not only extend the potent life of existing drug but also minimize the scope and expenditure of testing required for FDA approval and which make clinically already established drugs do their therapeutic best [14]. Therefore, the usage of novel drug delivery systems is a logical approach to circumvent these problems and effectively treat the HIV infection.
Controlled drug delivery system appeared to be the most effective drug delivery system in order to fulfil the need of a long-term treatment with antiretroviral agents [4]. The main shortcomings in conventional dosage forms are frequent administration, low water solubility, drug-plasma concentration fluctuations, poor bioavailability and significant adjustment in the life style. These drawbacks necessitated to develop controlled release drug delivery systems to improve the overall therapeutic benefit of anti-HIV drugs and to achieve effective therapy [15]. Bio-adhesive drug delivery systems are designed for prolonged retention on the mucosa to facilitate drug absorption over a prolonged period of time by interacting with mucin. Novel drug delivery carriers forms such as nanoparticles, liposomes, microparticles and others, has the advantage of overcoming the pharmacokinetic hurdles of anti-HIV drugs [16]. A study reported that administration of oral controlled release dosage forms leads to longer gastric residence time; lower the dosing frequency and constant maintenance of blood –drug levels [17]. A research involved on controlled release matrix tablets of zidovudine found; controlled release tablets with pH independent drug release characteristics and an initial release of 17 - 25% in first hour and extending the release up to 16 – 20 hours, can overcome the disadvantages of conventional tablets of zidovudine [18]. Another study of lamivudine oral controlled release tablets found the release of 20 - 30% drug in the first hour and extend the release up to 16 to 20 hours, can overcome the disadvantages associated with conventional tablet formulations of lamivudine [19].
Transdermal drug delivery system releases the drug by zero (or pseudo zero order) or by first order or both kinetics and which maintain the drug level for prolonged period for desired action. Transdermal delivery of anti HIV agents improve the bioavailability, reduce dosing frequency, avoid side effects and hepatic first pass metabolism which are the most common problems faced with the oral route [20].
Treatments for AIDS and HIV with conventional Antiretroviral drugs cause a lot of side effects due to its high dose and first pass metabolism. Due to that a it is suitable to give another route like trandermal route that reduces side effects and its dose. The absorption of drugs through the transdermal route improves bioavailability of drugs that might otherwise be metabolized by first-pass effect (pre- systemic drug elimination) during their passage through the gastrointestinal tract. Drug absorption from the transdermal route is mainly via passive diffusion through the lipoidal membrane. Thus, transdermal route of drug delivery has attracted the attention worldwide for optimizing the drug delivery [20]. To optimize the drug bioavailability, to overcome the side effects and prevent taste-related avoidance, several research groups have explored the potential of the transdermal route for the delivery of NRTIs. In a pioneering work, Pokharkar, et al. investigated the Effect of penetration enhancers on gel formulation of Zidovudine: In vivo and ex vivo studies through rat abdominal skin [21]. Nripendra, et al. investigated the effect of Skin Permeation of Zidovudine from Crosslinked Chitosan Film Containing Terpene Enhancers for Transdermal Use. through Albino rat [22]. Singh, et al. investigated the Effects of penetration enhancers on in vitro permeability of zidovudine gels [23]. Nripendra, et al. studied the Improvement of Skin Permeation of Zidovudine with Penetration Enhancers: In vivo – In vitro Correlations [24]. Pandey, et al. studied Transdermal delivery of stavudine using penetration enhancers [25]. From these above studies researchers was found that achieving therapeutically effective plasma concentrations would be possible with these formulations with avoiding side effects and improving bioavailability of drug candidate.
Micro particulates are small solid spherical particles within the size range of 1-1000μm [26]. Based on the method of preparation, the drug is either dissolved or entrapped, and encapsulated to the micro particle matrix. Micro particles provide easy administration to deliver macromolecules by various routes and effectively control the release of drugs over the periods ranging from few hours to months. Effective protection of encapsulated drug by various polymers prevents its degradation in the body. It is important in novel drug delivery system as they are prepared for controlled drug delivery which improves bioavailability of the drug and to target the specific sites in the body [27- 30].
Mucoadhesive microspheres include microparticle and microcapsules (having a core of the drug) of 1-1000 μm in diameter and consisting either entirely of a mucoadhesive polymer or having an outer coating of it, respectively [31]. Microspheres, in general, have the potential to be used for targeted and controlled release drug delivery, but coupling of mucoadhesive properties to microspheres as additional advantages, e.g. efficient absorption and enhanced bioavailability of the drugs due to high surface to volume ratio, a much more intimate contact with the mucus layer, specific targeting of the drug to the absorption site achieved by anchoring plant lectins, bacterial adhesives and antibodies on the surface of the microspheres [32]. Mucoadhesive microspheres can be tailored to adhere any mucosal tissue including those found in eye, nasal cavity, urinary and gastrointestinal tract, thus offering the possibilities of localised as well as systemic controlled release of drugs [33].
Lentiviruses of HIV, are able to infect all structures in the nervous system. It is able to invade the CNS by means of peripherally infected leukocytes, mainly monocytes and targets brain mononuclear macrophages, perivascular macrophages and microglia [34,35]. Enter the CNS compartment from the systemic circulation via two routes: i) through the blood-cerebro spinal fluid barrier (BCSFB) at the choroid plexus as cell-free viral particles and/or ii) through the blood-brain barrier (BBB) in form of infected monocytes [36,37]. The second route is known as the “Trojan horse approach”. In brief, monocytes infected by HIV-1 are able to cross the BBB between the capillary endothelial cells in a complex process regulated by the secretion of chemokines (e.g. MIP-1a/b, MCP-1, RANTES) from glial cells [38]. The brain macrophages and microglial cells, upon infection are responsible for further production of HIV-1 virus, and can also release viral proteins such as glycoprotein 120 (gp120), Tat (transcriptional activator) and Vpr (viral protein R) [39-42]. These viral proteins have been shown to be neurotoxic in vitro and trigger various harmful events such as activation of apoptotic pathways, cell-cycle arrest of neuronal cells and stimulation of the production of reactive oxidative species, glutamate, cytokines and other inflammatory factors from uninfected astrocytes which further accelerate the neurodegeneration process [43-45].
To effectively treat HIV-associated CNS complications, it is highly critical to improve the efficiency of CNS penetration by ARVs. The share of the administered dose of the drug that can reach the brain is consequently quite limited. The CNS penetration by ARVs is further compromised by the presence of the BBB and BCSFB [46]. BBB and BCSFB are equipped with specialized anatomical structures which dramatically prevent access of several exogenous compounds to the CNS compartment [47,48]. It is obvious that in addition to the anatomical features, the BBB and BCSFB have other mechanisms to control passage of drugs into the CNS. Indeed, there are several membrane transporters located at these two barriers which mediate their efflux from the CNS compartment back to the blood. Most of these transporters belong to the super family of ATP-binding Cassette (ABC) membrane transporters [49]. Furthermore, cerebral blood flow and degree of local inflammation can also affect drug CNS permeability. A full ABC-transporter consists of two transmembrane domains and two ATP-binding domains, whereas a half transporter consists of only one of each [50]. Several ABC transporters, specifically P-glycoprotein (P-gp), multidrug resistance-associated proteins (MRP) isoforms, and ABCG2 are known to be involved in the cellular extrusion of a broad range of drug molecules. P-gp, a full transporter also known as MDR1 protein, ABCB1 or CD243, is probably the most studied and characterized ABC member. It was first found as a 170-kDa ATP dependent membrane glycoprotein that acts as a drug efflux pump [51]. Many ARVs are large, lipophilic compounds with fairly high molecular weights. They are therefore likely candidates to be ABC transporter substrates. Several studies in polarized and P-gp overexpressing cell lines have indeed shown that various PIs (e.g. amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir) are substrates of P-gp [52-56]. A few recent studies have further demonstrated that NRTIs and NNRTIs are also capable of interacting with ABC membrane transporters. For instances, delavirdine is a P-gp blocker, whereas abacavir and stavudine are substrates of MRP4 and MRP5, respectively [57-59]. There are other lines of evidence supporting the frequent interactions of ABC transporters and ARV compounds. Therefore, strategies enhancing antiretroviral drug levels and distribution in the CNS are required.
ARVs can be effectively delivered to the brain using drug carriers of nanometer or submicron scale. Depending on the type of nanocarriers, chemical modifications of ARVs are often not required for efficient loading and delivery.
Nanocarriers can be classified according to the material used for their manufacture, such as liposomes, dendrimers, polymeric nanoparticles, solid lipid nanoparticles, and metal nanoparticles. They are also classified as nanospheres, nanocapsules, or nanoparticles based upon the dispersion of drug within the nanocarrier. Different types and modifications of nanocarriers have been studied for improved delivery of antiretroviral drugs. The use of nanocarriers can improve brain delivery of ARVs in several ways. Many of the agents delivered are well-established substrates of ABC transporters. These include P-gp substrates, e.g. doxorubicin, digoxin, rhodamine, vinblastine, and MRP substrates, e.g. methotrexate, fluorescein [60,61]. It is believed that with significantly higher levels of ARVs reaching the CNS, it is possible to reduce their doses and shorten the length of therapy. For example, the dosing frequency can be reduced by using a carrier that releases the ARV in a sustained manner. The circulation time can be prolonged and non-specific tissue binding reduced by coating a nanocarrier with polyethylene glycol (PEG) [62]. These are discussed in the following sections.
Nanocarrier-mediated drug delivery across the BBB can enhance brain delivery by three major pathways, which include: i) increasing the local drug gradient at the BBB by passive targeting, ii) allowing drug-trafficking by non-specific or receptor-mediated endocytosis and iii) blocking drug efflux transporters at the BBB. In addition to simply staying on the endothelium surface to release the loaded drug, some nanocarriers can enter cells by endocytosis, a pathway that allows “drug-trafficking” [63]. This can occur via nonreceptor mediated or receptor-mediated mechanisms. One example of the non-receptor mediated endocytosis is macropinocytosis. Macropinocytosis is a relatively non-specific processwhich allows cellular uptake of large particles up to the micron size range [64]. This is likely a useful pathway for nanocarriers such as solid lipid nanoparticles, which are frequently 200 to 300 nmin diameter. Macropinocytosis is also related to the uptake of Tat-peptides [65]. Current use of nanocarriers for brain delivery of ARVs have been studied in vitro or animal models [15,66-68]. These nanocarriers generally fall into a few broad categories: polymer/dendrimer-based, lipid-based or micelle-based.
Dendrimers are a versatile class of regularly-branched macromolecules with unique structural and topologic features. Small size (typically less than 100 nm), narrow molecular weight distribution, and relative ease of incorporation of targeting ligands make them attractive candidates for drug delivery. Dendrimers have minimal polydispersity and high functionality. Dendrimers can not only act as carriers of antiretroviral agents, but can also themselves act as antiretrovirals. Dendrimers with inherent antiretroviral activity can be synthesized by incorporating certain functional groups on their surface that can interfere with the binding of the virus to the cell. Dendrimers primarily act by blocking viral fusion to target cells and thus act as entry inhibitors in the early stages of viral infection, although secondary mechanisms of action at later stages of the viral life cycle have been reported [69-71]. Water-soluble dendrimers can be used as efficient carriers of antiretroviral agents which can be entrapped in the dendrimer architecture. The antiretrovirals or their prodrugs can also be grafted covalently onto the surface of the dendrimers, either alone or in conjunction with other molecules, such as targeting moieties and fluorescent tags. Multivalent dendrimeric systems have been of much interest in the field of antiviral therapy. Dutta, et al. developed efavirenz-loaded, tuftsin-conjugated poly(propyleneimine) dendrimers for targeted delivery to macrophages [69,72]. These multivalent dendrimers showed reduced cytotoxicity compared with nonconjugated. Poly(butyl cyanoacryalate) (PBCA), an acrylic polymer, has so far been the most studied polymer for brain delivery. PBCA nanoparticles have demonstrated good accumulation in both brain tissues and cerebrospinal fluid without physical disruption of the BBB integrity [73]. PBCA is biodegradable and its lipophilicity facilitates efficient encapsulation of diverse types of neutral and weak base compounds such as dalargin, loperamide, amitriptyline, methotrexate and doxorubicin [73-75]. Charged acrylic co-polymers such asmethylmethacrylate-sulfopropylmethacrylate (MMASPM) were therefore studied as a substitute. Their negative charges grant them a higher loading capacity for polar compounds including zidovudine when compared to PBCA [76]. This MMA-SPM nanocarrier system was able to increase the permeability of zidovudine and lamivudine across an in vitro BBB model of bovine brain-microvascular endothelial cells by 8–20 and 10–18 folds, respectively [76]. Polyesters such as poly (D,L-lactide-co-glycolide) (PLGA) and polylactide (PLA) are two of the few polymers officially approved for clinical use. Their molecular weights, hydrophilicity, degradation rate and hence the release kinetics can be conveniently tailored by adjusting the composition [77]. They also easily form hydrolysable bonds with diverse therapeutic molecules and targeting moieties such as lectin [77-79]. Drug loading into PLGA/PLA nanocarriers and modification of these systems for brain targeting are therefore quite convenient. Use of PLGA/PLA based nanocarrier specifically for ARVs delivery to brain has not been reported. This is clearly an area for further study. Like regular polymers, dendrimers also consist of repeating monomer units, but dendrimers are characterized by their repeatedly branched molecular structures. These highly branched molecules,when precisely engineered, can form mono dispersed globular or spheroidal nanostructures of 1 to over 10 nm in diameter [80]. They have been shown to increase the BBB permeability of therapeutic agents such as DNA and methotrexate [81,82]. Recently dendrimers have been evaluated for CNS delivery of ARVs. Polyamidoamine dendrimers loaded with lamivudine, a NRTI commonly used in HIV treatment, were evaluated for their in vitro antiviral activity in MT2 cells infected with HIV-1. When loaded on dendrimeric nanocarriers, a 21-fold increase in cellular lamivudine uptake and 2.6-fold reduction in the viral p24 levels were observed when compared to the group treated with free drug solution [83]. Despite these promising results, it should be noted that the drug release kinetics of dendrimers are sometimes in consistent, and their long-term safety profiles are relatively less established than polymers like PLGA. More in vivo data are needed to further validate the use of dendrimeric nanosystems for ARVs delivery to the CNS.
A number of lipid-based formulations (e.g. liposome, lipoplex) are already commercially available and all of them have solid track record of clinical safety. There are several classes of lipid based nanocarriers available, including liposomes, micro- or nanoemulsion and solid lipid nanoparticles (SLN). Liposomes are regarded as an inert carrier system having lipophilic tails oriented to the middle and polar head directed to inside of the vesicle and its outer surface. These vesicles are able to encapsulate both hydrophilic and lipophilic active ingredient in their structure and protect them from degradation [84,85]. A variety of natural and synthetic phospholipids are available for the preparation of liposomes. Because of their entrapping ability, liposomes are being considered as drug-carrying structures or vesicles. When materials are encapsulated in a liposome, the oil soluble actives sequester in the phospholipid bilayer (tail), while the water-soluble actives reside in the water core (head). These spherical vesicle with an aqueous cavity at their center are either unilamellar, having single bilayer or oligolamellar with multiple bilayers or multilamellar with a large number of bilayers. Liposomes are either homogeneous with a narrow distribution or heterogeneous with a broad distribution. The size of a liposome ranges from some 20 nm up to several micrometers and they may be composed of one or several concentric membranes, each with a thickness of about 4 nm. A schematic picture of a liposome is shown in Figure 1.
Liposomes in their native form are taken up by the reticuloendothelial system and are quickly cleared from the circulation. This property was exploited for the macrophage delivery of antiretrovirals. Phillips, et al. used a liposomal drug delivery system to deliver zidovudine to macrophages [69,86]. Zidovudine, which is an amphiphilic compound, showed low entrapment and significant leakage from the liposomal vesicles over time, due to its tendency to partition between the aqueous core and the lipid bilayer [86,87]. Jin, et al. studied the incorporation of the myristate prodrug of zidovudine in the liposomal formulation to target macrophages [88]. The prodrug, being lipophilic, resulted in greater entrapment efficiency and a longer half-life, as demonstrated in vivo in rats [86]. A number of liposomal systems have been developed and evaluated for the treatment of various brain illnesses, such as cerebral ischemia by citicholine brain tumors by cisplatin and epilepsy by phenytoin [89-91]. Overall, significant improvement in brain drug levels were observed in these studies. Although there are a few liposomal formulations for delivery of ARVs, e.g. stavudine and zidovudine relatively few of them are designed for HIV associated CNS illnesses [92,93]. Foscarnet is an antiviral used as a salvage therapy for late-stage HIV patients with multidrug resistance. Liposomal foscarnet was able to increase the drug level in rat brains by 13-fold when compared to the free foscarnet solution [94]. Another drug, amphotericin B, is commonly used to treat the opportunistic fungal infections in HIV patients. However, amphotericin B does not cross the BBB. The use of liposomes coupled with brain targeting peptides for amphotericin B delivery significantly increased the drug transport across the simulated BBB model formed by rat brain endothelial cells [95]. These studies further support the use of liposomes for more effective treatment of HIV-associated CNS complications.
Colloidal drug delivery systems such as liposomes and niosomes have distinct advantages over conventional dosage forms. These systems can act as drug reservoirs and provide controlled release of the active substance. In addition, modification of their composition or surface can allow targeting [96]. Niosomes are non-ionic surfactant based vesicles that had been developed as alternative controlled drug delivery systems to liposomes in order to overcome the problems associated with sterilization, large-scale production and stability. Colloidal drug carriers (niosomes) are easily phagocytosed by macrophages. Therefore, they can facilitate the uptake of antiviral drugs by these cells and may enable a considerably improved AIDS therapy. Niosomes formed from self-assembly of hydrated synthetic nonionic surfactant monomers capable of entrapping variety of drugs [96]. The size of these vesicles is in the nanometer range. This size range offers the decisive advantage of this class of pharmaceutical dosage forms as it allows drug targeting which often is not possible with free drug.
Nanoemulsions and microemulsions are usually oil-in-water formulations in which the oil phase is highly dispersed to droplets of submicron size and stabilized by surfactants and co-surfactants. This type of formulations is especially suitable for highly lipophilic HIV drugs such as PIs. Recently, saquinavir, the first PI marketed for HIV treatment, was evaluated for brain delivery in an oral formulation of flaxseed oil-based nanoemulsion [97]. The average oil droplet size was around 100 to 200 nm in diameter. Use of saquinavir nanoemulsion instead of its free drug solution resulted in a three-fold increase in the saquinavir concentration in the systemic circulation and three and five-fold increase in the area-under-the curve (AUC) values. This study shows that in addition to enhancement of BBB permeability, the small size of the nanoemulsion may also help bypass other barriers such as the gastrointestinal tract when used as oral formulations. SLN are a relatively new class of lipid-based nanocarriers [98]. They are made of one or more lipids with melting points higher than body temperature, so the carriers remain in solid state after administration. The low solubility of nanocarrier biomaterials probably contributes to the high tolerability of this formulation. A study showed that SLN in fact caused less non-specific cell toxicity even compared to nanoparticles made of PLGA which has long been the standard for biocompatible materials [99]. In addition, by getting immobilized within a lipophilic environment, the loaded drugs can be more adequately protected from degradation. SLN evidently hold strong promise for brain delivery of ARVs, especially PIs which are mostly lipid soluble.
A micelle is an aggregate formed by typically 50–100 amphiphilic molecules (e.g. surfactants, block-copolymers) when dispersed in a liquid phase [100]. In aqueous solution the amphiphilic molecules aggregate and expose their hydrophilic heads outside and hide their hydrophobic segments in the inner core region. This structure facilitates solubilization of hydrophobic drug compounds within the micelle core. The size of a micelle usually falls in the range of 5 to 20 nm in diameter. The small size and good drug solubilization properties make micelles potentially valuable nanocarriers. Pluronic micelleswere shown highly effective for BBB drug transport enhancement in vitro and in vivo [100-102]. Using bovine brain microvessel endothelial cell monolayer model, the effect of Pluronic P85 on the permeability of a broad range of structurally unrelated compounds was examined by Kabanov’s group [102,103]. Increases in the drug permeability up to 19-fold were detected. This permeability enhancement was particularly strong with P-gp substrates such as paclitaxel, vinblastine as well as ritonavir a PI frequently used in HAART regimens as a booster [103]. In animal models, Pluronics increased the drug delivery to the brain of wild-type mice, but the same benefit was not observed in mdr1a/b knockout mice, indicating that the drug permeability enhancing effect by Pluronics ismediated at least in part by P-gp inhibition at the BBB [100]. It was suggested that this P-gp suppressive effect could be mediated by ATP depletion, membrane fluidization by the co-polymer, or a combination of both mechanisms [102-104].
Nanoparticle formulations help in various health challenges in treating diseases like cancer, psychiatric, neurological disorders and vaccines, protein therapeutics and gene therapy. The following are a few advantages of nano technology for drug therapy like passive targeting of drug involves nano particles targeted (cellular/tissue) delivery of drugs. Considering the serious problems associated with HIV therapy, the anti-retroviral drug delivery development like nanoparticle based formulations will help us in making anti-retroviral therapy more effective. This will overcome the problems of drug crossing the biological barriers and to reach the affected site more effectively. This will also overcome the bioavailability problems of the drug. Nanotechnology involves assembling individual atoms or molecules in-to useful nanoscale structures. However, it is very hard to do with metals in general because they oxidize rapidly under atmospheric conditions. For example, a nanoparticle of iron becomes a particle of iron oxide rust in only a few seconds. Although some metals can be protected for a while by the formation of a thin oxide film, it’s better to use metals that are inert in the atmosphere such as gold. As a result, gold particles, wires and surfaces are at the heart of much of nanotechnology. Resistance to corrosion, electrical conductivity and special affinity for sulfur-containing organic molecules are particularly attractive features of gold nanoparticles. These properties have lead to quite interesting and promising structures, such as ultra-sensitive biosensors [105]. One of the features of gold and silver nanoparticles is that they possess a range of quite unusual colors. Bulk gold has a familiar yellow color, which is caused by a reduction in the reflectivity of light at the blue end of the spectrum. However, if we subdivide gold into smaller and smaller particles, there comes a point at which the particle size becomes smaller than the wavelength of incident light. New modes of interaction between the radiation and the gold become prominent, in particular interactions involving electronic oscillations called surface plasmons. When the particles of gold are small enough, they are ruby red in color. This coloration is due to the gold particles strong absorption of green light, corresponding to the frequency at which a resonance occurs with the gold. This effect has been used to color glass, even as far back as Roman times. However, it is a transmission color; if the same nanoparticles are viewed in reflected light, they will appear orange or green-brown. This gives an interesting color-shift to translucent artifacts colored with metallic nanoparticles. These colors may also be readily varied by adjusting the shape or state of agglomeration of the metallic nanoparticles. A particularly useful color change is produced when discrete gold nanoparticles agglomerate into groups that resemble bunches of grapes. The color of a suspension or coating of such particles changes from burgundy to inky blue. This phenomenon has been invoked as a colorimetric indicator in home pregnancy tests and in testing for specific genetic sequences. This combination of properties has encouraged its use in a diverse range of niche applications [106]. Silver nanoparticles were shown to be effective against a wide range of HIV-1 strains in vitro, including laboratory strains, clinical isolates, M and T tropic strains, and resistant strains [86,107]. Silver nanoparticles act as viral entry inhibitors by binding to gp120 and thus preventing CD4-mediated viral membrane fusion to host cells and subsequent infectivity [107,108]. They are also found to inhibit post-entry stages of HIV-1, indicating that silver nanoparticles act at multiple stages of the HIV life cycle [86,107]. Another inorganic nanoparticulate system evaluated for antiretroviral drug delivery consists of gold nanoparticles. The use of gold colloid in biological applications began in 1971, when the immunogold staining procedure was invented. Since that time, the labeling of targeting molecules, especially proteins, with gold nanoparticles has revolutionized the visualization of cellular or tissue components by electron microscopy. The optical and electron beam contrast qualities of gold colloid have provided excellent detection qualities for such techniques as immunoblotting, flow cytometry and hybridization assays.
Synthetic or semisynthetic polymers, such as poly(lactic acid) (PLA), poly(lactic-co-glycolic acid) (PLGA), poly(alkyl) cynoacrylates, poly(ethylene glycol-co-(lactic-glycolic acid)), poly(caprolactone), and poly(methyl) methacrylate, are frequently used for the manufacture of nanoparticles intended for drug delivery. PLA and PLGA have been approved for human use by the Food and Drug Administration (FDA) [86]. The biodegradable and biocompatible properties of polymeric nanoparticles make them very attractive candidates for drug delivery, tissue engineering, and biomedical applications. When the drug particles are unstable physico-chemically and/or biochemically in a particular biological environment, e.g. pH (gastric juice), presence of enzymes, the solid nanoparticles can be converted to polymeric nanoparticulate systems in which the drugs are encapsulated in the form of a solid solution or dispersion or adsorbed on the surface or chemically bound. Such a drug– polymer hybrid or combined system offers the possibility of giving the loaded drug more sophisticated or tailor-made delivery properties. The particle size and distribution, surface charge, hydrophilicity and hydrophobicity can determine the functions of nanoparticles or nanospheres. There are many options for modifying these properties of nanospheres by selecting appropriate polymer materials, e.g. naturally occurring or synthetic. Biodegradable and biocompatible polymers, such as lactide glycolide copolymer are the most suitable [86,109].
Solid lipid nanoparticles (SLNs) are made of fatty acids that are solid or semisolid at room temperature. Lipids with high melting point such as stearic acid, glyceryl monostearate, cetyl palmitate, and hydrogenated cocoglycerides and emulsifiers, such as phosphatidylcholine, lecithins, polysorbates, poloxamers, bile salts, and fatty acid co-esters are used to prepare and stabilize the SLNs, respectively. Sustained drug release and site specificity for drug delivery can be achieved by altering the properties of SLNs, such as their lipid composition, size, and surface charge. This delivery system offers several advantages such as relative ease of production, sterilization, and scale-up, avoidance of the use of organic solvents, low-cost excipients, and biocompatibility. Compared with nanoemulsions which are liquid lipid encapsulations of the drug, SLNs containing the lipid in the solid state impart greater drug stability and better control over drug-release kinetics [86].
Various nanocarrier-mediated solutions have been proposed in order to increase antiretroviral drug delivery to the CNS. For example, the ability of nanostructured lipid carriers (NLCs) to potentially mediate the brain delivery of zidovudine, a frequently used nucleoside reverse-transcriptase inhibitor that has related toxic effects and a very short plasma half-life (around 1 hour), was studied in vitro by Joshy and Sharma [110]. Albumin is a matrix-forming macromolecule commonly used in the production of drug nanocarriers, mainly due to its advantageous biodegradability and low toxicity properties. Albumin NPs encapsulating zidovudine have been proposed for brain delivery of this polar nucleoside; alongside, surface modification of NPs with PEG was performed in order to reduce their rapid removal from blood circulation [111]. The NP surface was further modified by anchoring Tf ligand with the purpose of enhancing CNS uptake. The biodistribution of zidovudine was studied in Wistar rats after intravenous administration of unmodified and Tf-modified PEG-albumin NPs with an equivalent dose of 54.4 mg drug/kg of body weight. Experimental results showed significant enhancement of brain drug localization for Tf-PEG-albumin NPs compared to PEG-albumin NPs (21.1% ±1.8% versus 9.3% ±0.9%, respectively, of the total drug recovered at 4 hours postadministration). This study confirmed the potential use of Tf modification of nanocarriers in order to enhance brain drug targeting for HIV therapy. In order to target macrophages, either at the CNS or the mononuclear phagocyte system, Kaur, et al. developed didanosine-loaded gelatin NPs with mannan coating [112]. These macromolecular (gelatin) NPs were produced by double desolvation, and in order to coat them, they were incubated with a mannan solution. Besides providing sustained drug release, this nanosystem improved in vitro drug uptake by macrophages, with a five fold increase in cell-associated levels after 2 hours’ incubation compared with didanosine in solution. Fluorescence microscopy also confirmed NP cell internalization. Further, didanosine levels in the brain were increased by 12.4-fold upon subcutaneous administration of mannan-coated NPs compared to didanosine in solution (Figure 1) [112]. Also, brain accumulation of didanosine was higher than when plain NPs (ie, without mannan surface modification) were used. These results seem to support the positive influence of mannose residues in the ability of NPs to reach the CNS, probably by a monocyte/macrophage-mediated mechanism. Poly(l-lactic acid) NPs conjugated with Tat peptide were also studied in order to increase the transport of ritonavir across the BBB and into the CNS [113,114]. This strategy was envisioned in order to bypass the known efflux action of P-gp on this particular PI. NPs were produced by an emulsion-solvent evaporation technique and Tat functionalization achieved by using an epoxy conjugation method, where the NP surface is activated by epoxy compound followed by Tat conjugation. NPs were shown to be effective in vitro in inhibiting HIV-1 infection of monocyte-derived macrophages through the reduction of cytopathic effects, HIV-1 p24 protein secretion, and production of progeny virions [114]. Also, P-gp intact (wild-type) mice were injected via the tail vein with either ritonavir-loaded NPs or the drug in solution (45 mg/kg ritonavir in both cases) [113]. Rao, et al. proposed that Tat-conjugated NPs were transported to the parenchyma without influencing the integrity of the BBB, which suggests that this transport could occur due to transcytosis across the endothelium of the brain vasculature. Moreover, these NPs maintained potentially therapeutic drug levels in the brain for a sustained period (14 days) with a single-dose intravenous administration [113]. Therefore, Tat-NPs may constitute an effective way of delivering anti-HIV-1 drugs to the CNS. In another approach, Mahajan, et al. proposed the use of Tf-conjugated quantum rods (QRs; average length and diameter of approximately 25 nm and 5 nm, respectively, as assessed by transmission electron microscopy) in order to enhance the brain delivery of saquinavir [115]. QRs were composedof a thin zinc sulfide layer over a cadmium/selenium core nanocrystal and coated with mercaptosuccinic acid; saquinavir and Tf were further conjugated to surface carboxyl groups by carbodiimide chemistry and stabilized by mixing with poloxamer 407. The nanocarrier was tested in an in vitro BBB model comprising primary cultures of HBMECs and normal human astrocytes in a double-chamber setup. An HBMEC monolayer was grown in the apical side of a porous polyethylene terephthalate membrane, while the basolateral side was covered with an astrocyte monolayer. Additionally, HIV-1-infected peripheral blood mononuclear cells (PBMCs) were placed in the receptor chamber in order to evaluate the antiretroviral efficacy of different treatments. Results showed that QRs did not affect the integrity of the BBB, and presented reduced cytotoxicity of up to 40 nM concentration in saquinavir; also, functionalization with Tf led to enhanced uptake of QRs by HBMECs and PBMCs compared to nonfunctionalized QRs. Moreover, when the above BBB model was used, a significant decrease of HIV-1 replication in PBMCs was observed for Tf-saquinavir QRs compared to both the free drug and non-Tf-functionalized saquinavir QRs. These observations seem to support the view that the proposed QRs may provide an efficient way to promote the transport of saquinavir across this barrier and reduce the viral load at the CNS. Nevertheless, the mechanism of release of conjugated drugs from the carrier is not clear, even though the authors claim that degradation of the nanosystem as a whole is involved [40]. Comparable results were also reported for Tf-conjugated QRs carrying amprenavir [116]. In addition, these QRs present the ability to be used in real-time tracking of particle crossing through the in vitro BBB model, as well as a potential use in diagnostic imaging, due to their intrinsic optical properties [117]. Of further note, nanoplexes have also been obtained by the same group by using quantum dots (QDs) produced with the same materials and presenting a similar structure as QRs (in this case, Tf was not conjugated and poly[diallyldimethylammonium chloride] was used to provide a positive surface charge to QDs) and siRNA targeting the expression of matrix-degrading metalloproteinase type-9 [118]. This protein plays a significant role in disrupting the BBB, and its expression is triggered by HIV-1 infection of the CNS. Results using the aforementioned in vitro BBB model showed that proposed siRNA–QD nanoplexes were able to silence gene expression in HBMECs and thus maintain the integrity of the membrane. Although not directly targeting the virus, this delivery system may provide an interesting auxiliary approach in preventing continuous viral invasion of the CNS. Added to the previous examples of different nanocarriers, size reduction to the low-micrometer/nanometer scale of antiretroviral drugs, and their eventual surface modification, may also be an interesting strategy of enhancing drug delivery to the CNS. For example, Shegokar and Singh prepared nanosuspensions of nevirapine by high-pressure homogenization and proceeded with surface modification with serum albumin, PEG 1000, or dextran 60 by physical adsorption in solution (final diameters around 500 nm) [119]. When tested in vivo in a rat model, particles modified with albumin presented the ability to accumulate in the brain compared to nevirapine in solution or other drug nanosuspensions (in the previous cases, no drug was detected at this anatomical site). The area under the curve in the brain (AUCbrain)/AUCblood ratio of albumin-modified nanosuspensions was 9.33. A recent study by Dash, et al. further demonstrated the potential of using nanosuspensions of atazanavir and ritonavir obtained by high-pressure homogenization and using poloxamer as a stabilizer for neuroprotection in a humanized HIV-infected animal model (nonobese diabetic/severe combined immunodeficiency-γcnull mice) [120]. Weekly intravenous administration of nanosuspensions allowed the eliciting of neuroprotective responses (as assessed by reduced neuronal, synaptic, and astrocyte damage), alongside the reduction of viral loads and maintenanceof CD4+ cells in peripheral blood. Indeed, monocytes/macrophages may be an ideal physiological “shuttle” for brain delivery of antiretroviral drugs, due to their phagocytic nature, which allows extensive uptake (depending on coating, size, shape, and charge) and sustained release (for days to weeks) of different antiretroviral drug particles [121-123]. Researchers observed that nanoformulated indinavir could be delivered into the brain of an HIV-1 encephalitis rodent model up to around 20-fold higher levels when incorporated into bone marrow-derived macrophages after a single dose administered intravenously [124]. Moreover, treatment with this cell-based nanoformulation was able to release indinavir in a continuous fashion over 2 weeks and reduce HIV replication in brain regions presenting signs of encephalitis. One important aspect of using monocytes/macrophages for the delivery of nanosized antiretroviral drugs to the CNS is related to the potential of drug-induced toxicity to cell carriers.
Acquired Immuno Deficiency Syndrome (AIDS), which threatens to cause a great plague in the present generation. It is crucial for the success of AIDS therapy to maintain the systemic drug concentration consistently above its target antiretroviral concentration throughout the course of the treatment. Conventional drug delivery involves the formulation of the drug into a suitable form, such as compressed tablet for oral administration or a solution for IV administration. These dosage forms have been found to have serious limitations in terms of higher doses required lower effectiveness, toxicity and adverse effects. NDDS are being developed rapidly, so as to overcome the limitations of conventional drug delivery. To minimize drug degradation and loss, to prevent harmful side-effects and to increase drug bioavailability and the fraction of the drug accumulated in the required zone, various drug delivery and drug targeting systems are currently under development as a means to enhance the effective delivery of antiretroviral drugs for HIV prevention and therapy. Among drug carriers one can name soluble polymers, micro particles made of insoluble or biodegradable natural and synthetic polymers, microcapsules, cells, cell ghosts, lipoproteins, liposomes, niosomes and micelles. The carriers can be made slowly degradable, stimuli-reactive (e.g., pH- or temperature-sensitive), and even targeted (e.g., by conjugating them with specific antibodies against certain characteristic components of the area of interest). Targeting is the ability to direct the drug-loaded system to the site of interest. The mode of delivery can be the difference between a drug’s success and failure, as the choice of a drug is often influenced by the way the medicine is administered. Sustained (or continuous) release of a drug involves polymers that release the drug at a controlled rate due to diffusion out of the polymer or by degradation of the polymer over time. Different strategies have been proposed to further improve the various aspects of this novel therapeutic approach, from efficiency, safety and specificity. The physicochemical parameters of nanocarriers such as their size, surface charge and hydrophilicity can be optimized to favor the nonspecific, passive form of brain targeting. The authors suggested that particles smaller than 100 nm could better mimic the membrane receptors (e.g. low density lipoprotein receptors) and be more efficiently transcytosed via receptor-mediated pathways. For specific brain targeting, various biomolecules expressed at the BBB can be typically fall into two categories — indirect targeting and direct receptor targeting. With indirect targeting, nanocarriers are made of materials that bind to specific molecules in human body, which have high affinity with the receptors at the BBB. With direct targeting, nanocarriers are surface-grafted with ligand molecules that specifically target those receptors at the BBB. Polysorbates (also known as Tweens) are a commonly used class of non-ionic surfactants. They have very low toxicity and are officially approved for intravenous use. Overall, the potential drawback of this class of specific receptortargeting platforms lies in their cost. To obtain large quantity of monoclonal antibodies of clinically useful grade at sufficiently low cost poses a major obstacle. Thus, various drug delivery and drug targeting systems has attracted the attention worldwide for optimizing the drug delivery. The review embracing the need for new drug delivery and recent development in drug delivery of anti HIV drugs for better management of life threatening disease. Among the recent approaches of novel drug delivery system for anti-HIV drugs, controlled/sustained and targeted/intracellular drug delivery are the important ones.

![]()

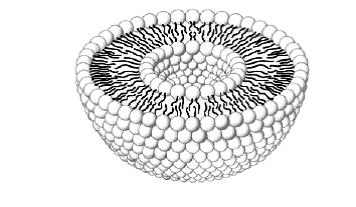 |
| Figure 1: Liposome structure |
DRUG FAMILY |
DRUG |
FDA APPROVA L DATE |
DOSAGE FORMS |
ADULT DOSE |
F % |
h |
| Nucleoside reverse transcriptase inhibitors (NRTIs) |
Zidovudine (AZT) |
19th March 1987 |
Capsule, tablet, syrup, injection |
600 mg/day (2 doses) |
60 |
1.1 |
Didanosine (ddI) |
9th October 1991 |
Tablet, solution |
400 mg/day (1 dose) |
30 to 40 |
1.3 to 1.6 |
|
Zalcitabine (ddC) |
19th June 1992 |
Tablet |
2.25 mg/day (3 doses) |
85 |
1 to 3 |
|
Stavudine (d4T) |
24th June 1994 |
Capsule, powder |
80 mg/day (1 or 2 doses) |
80 |
1 to 1.6 |
|
Lamivudine (3TC) |
17th November 1995 |
Tablet, solution |
300 mg/day (1 or 2 doses) |
86 |
3 to 6 |
|
Abacavir |
17th Dec. 1998 |
Film, coated tablet |
600 mg/day (1 or 2 doses) |
83 to 100 |
1 to 2 |
|
Emtricitabine |
2nd Oct 2003 |
Capsule |
200 mg/day (1 or 2 doses) |
93 |
10 |
|
Nucleotide reverse transcriptase inhibitors (nRTIs) |
Tenofovir |
26th Oct 2001 |
Tablet |
300 mg/day (1 dose) |
25 to 39 |
17 |
Non - Nucleoside reverse transcriptase inhibitors(NNRTIs) |
Nevirapine |
21st June 1996 |
Tablet |
Ist 14 days – 200 mg/day After 14 days – 400 mg/day (2 doses) |
90 |
25 to 30 |
Delavirdine |
4th Apr 1997 |
Tablet |
1200 mg/day (3 doses) |
NA |
5.8 |
|
Efavirenz |
17th Sept 1998 |
Capsule , film, coated tablet |
600 mg/day (1 dose) |
42 to 80 |
40 to 50 |
|
Etravirine |
18th Jan 2008 |
Tablet |
400 mg/day (2 doses) |
NA |
30 to 40 |
|
Protease Inhibitors (PIs) |
Saquinavir |
6th Dec 1995 |
Capsule |
2000 mg/day (2 doses) |
NA |
1.5 to 2 |
Indinavir |
13th Mar 1996 |
Capsule |
2400 mg/day (3 doses) |
65 |
1.2 to 2 |
|
Ritonavir |
1st Mar 1996 |
Capsule, solution |
1200 mg/day (2 doses) |
65 |
3 to 5 |
|
Lopinavir |
15th Sept 2000 |
NA |
NA |
NA |
4.4 |
|
Nelfinavir |
14th Mar 1997 |
Tablet, powder |
2500 mg/day (2 or 3 doses) |
20 to 80 |
3.5 to 5 |
|
Amprenavir |
15th Apr 1999 |
Capsule, solution |
2400 mg/day (2 doses) |
NA |
7 to 10 |
|
Fosamprenavir |
20th Oct 2003 |
Tablet |
2800 mg/day (2 doses) |
NA |
7.7 |
|
Atazanavir |
20th June 2003 |
Capsule |
400 mg/day (1 dose) |
NA |
7 |
|
Tipranavir |
22nd June 2005 |
Capsule |
1000 mg/day (2 doses) |
30 |
5.5 to 6 |
|
Darunavir |
23rd June 2006 |
Tablet |
1200 mg/ day (1or 2 doses) |
37 |
15 |
|
Virus fusion/entry inhibitors |
Enfuvirtide |
2003 |
Powder & Injectables |
180 mg/day (2 doses) |
84.3 |
3.8 |
Maraviroc |
2007 |
Tablet |
1200 mg/day (2 doses) |
23 to 33 |
14 to 18 |
|
Vicriviroc |
Phase III |
Tablet |
30 mg/day |
NA |
NA |
|
Integrase Inhibitors |
Raltegravir |
2007 |
Tablet |
800 mg/day (2 doses) |
NA |
9 |
Elvitegravir |
Phase III |
NA |
150 mg/day |
NA |
NA |
|
Maturation Inhibitor |
Bevirimat |
Phase II |
Tablet & Solution |
600 mg/day |
NA |
NA |
| * F – Bioavailability * h – Half - lives Table 1: Antiretroviral drugs, their commercially available dosage forms, bioavailabilities and half-lives [4,8-12] |
||||||




































