Open Access
Research Article
Max Screen >>
ISSN: 2348-9790
Copyright: © 2017 DebRoy C. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
In animal production, antibiotics are used at sub-therapeutic and therapeutic levels for growth promotion and disease prevention. Using antibiotics can result in selecting for drug resistant microorganisms that may spread to humans through consumption of contaminated food. Lately, an increased interest in organic farming, where animals are grown without antibiotics, has been popular amongst the consumers. It is believed that organic farming will reduce antimicrobial resistance in microorganisms. However, the animals grown organically may be prone to diseases, and other microorganisms may thrive in the gut, that may be potentially harmful to humans. We analyzed the DNA of fecal samples of pigs grown under conventional and organic dietary regimens and detected the presence of genes that causes antibiotic resistance. The microbiomes of gut in swine grown under these two dietary regimens exhibited little difference in composition and exhibited the presence of Bacteriodetes, Firmicutes and Proteobacteria at the phyla level and Prevotella, Lactobacillus, Barnesiella, Proteus, Fecalibacteria, Escherichia coli and Shigella at the genus level. Presence of eighty seven antibiotic resistance genes, tested by real-time PCR, exhibited the presence of more antibiotic resistance genes in guts of swine grown in conventional diet than under organic conditions. This pilot study may lead to comprehensive research on the composition of microbiomes in pigs in large production environment allowing formulation of strategies that may eventually reduce prevalence and dissemination of antibiotic resistance genes in the guts of animals.
Keywords: Swine Ricrobiome; Antibiotic Resistance Genes; Phylotypes; Metagenomic Library
Antibiotics have been administered at sub-therapeutic and therapeutic levels for animal production for many years in the United States for disease prevention and growth promotion. It has been demonstrated that antimicrobial resistance in foodborne pathogens may arise due to use of antimicrobials in agriculture that may spread to humans through contaminated food [1-4]. To address the concern, United States Department of Agriculture (USDA) established the National Organic Program in 1990 that has grown by almost 20% annually. Organic production focuses on animal health, good environmental practices, production quality and do not use antibiotics, hormones or animal byproducts in feed as mandated by USDA. (https://www.usda.gov/wps/portal/usda/usdahome?navid=organic-agriculture). Conventional farming practices generally focus on reducing costs and maximizing production by feeding antimicrobials and dietary supplements to animals.
Antibiotics in the diet plays a major role in modulating gut microflora by providing selective pressure on microbial community leading to the emergence of drug resistant bacteria. The selection pressure may also facilitate persistence and the transfer of resistance determinants between bacterial species [5,6]. It is now known that non-pathogenic commensal microorganisms may develop antibiotic resistance and also transmit the resistance determinants to other microbes [7,8]. The emergence and dissemination of antibiotic resistance in bacterial pathogens is a serious emerging problem worldwide [7,9]. In swine production, association between antimicrobial use and antimicrobial resistance is well documented [10-14]. Antimicrobial resistant bacteria are consumed by humans through contaminated food, or can be acquired by direct contact with animals, or from the environment due to release of contaminated animal waste [15]. To prove that antimicrobial usage in swine production may be directly correlated with antimicrobial-resistant infections in the human population has been challenging. However, diseases in humans, caused by multidrug-resistant Salmonella entericaserotype Typhimurium DT104 in Denmark [16], S. enterica serotype Typhimurium DT104 in England [17], and Enterococcus faecium in China [18] were found to have originated from swine herds.
In this investigation, we examined the differences in microbiomes of pigs grown under conventional and organic dietary regimens and analyzed the phylotypes associated with the gut microbiomes. The fecal samples were further enriched in selective media to examine the differences between the non-cultured and enriched fecal samples from organically and conventionally grown pigs. While the analysis exhibited that certain microorganisms were more prevalent in conventionally grown swine versus those that were organically grown, the differences were not significant. Distribution and expression of antibiotic resistance genes in microbiota in the gut of swine produced in conventional and organic farms exhibited some differences. More resistance genes were observed in pigs grown in the presence of antibiotics as compared to the ones grown organically. This investigation provides information on the type of microbes and the presence of antimicrobial resistance genes associated with gut microbiomes in pigs grown under organic and conventional dietary regimens that may potentially assist in defining and formulating diets for swine production.
Fresh fecal samples from pigs (5-6-weeks old) grown in organic (n=10) and conventional (n=10) dietary regimens were collected. The organic diet regimen contained crude protein (20.0%), crude fat (6.0%), crude fiber (4.3%), calcium (0.5-1%) and phosphorus (0.6%) (AN PRESTART 13-18). The samples were collected from the pigs after they were on this ration for 11-15 days following weaning for 3 weeks. Pigs grown in conventional diet were on ASP250 ration where the active ingredients were chlortetracycline (100 g/ton), sulfamethazine (100 g/ton) and penicillin (50 g/ton) at sampling. They were fed this diet from 3-5 days before sampling. For the first 9-11 days, after weaning, they were on PreStart 13-15XT DEN/CTC Medicated comprised of the main ingredients crude protein (20.3%), crude fat (6.0%), crude fiber (3.9%), calcium (0.5%-1.0%) and phosphorus (0.6%). The active drug ingredients were tiamulin hydrogen fumarate chlortetracycline (35 g/ton), chlortetracycline (400 g/ton). The rationale for using samples from pigs at this age was because the presence of antimicrobial resistance genes was expected to be apparent after feeding the diet at least for 11-15 days following weaning.
Fecal samples (1 gm) from each pig (n=20) were grown in 50 mL of Tryptic Soy Broth (TSB) overnight at 37 oC for enrichment.
DNA was extracted from the fecal samples from each of the 10 pigs grown in conventional or organic diet using the PowerMax fecal DNA Isolation Kit (MoBio Laboratories Inc. Carlsbad, CA) according to the manufacturer’s protocol. DNA extracted from each group, directly from the fecal samples or after enrichment in TSB was pooled and the concentration of total genomic DNA isolated from each group was estimated using Qubit® Fluorometer (Invitrogen, Molecular Probes, Eugene, OR). Pooled DNA (0.1 μg) from each group were used for amplification of 16S ribosomal RNA genes (rRNA) using universal bacterial 16S primers (F-5’GGAGGCAGCAGTRRGGAAT; R-5’CCCCGTCAATTCMTTTGAGTTT that amplified the V3 region. Sequencing studies focusing on hypervariable regions V3-V5 of 16S rRNA gene exhibited substantial taxonomic information that allows for discrimination similar to whole genome sequencing [19]. V3 region was therefore selected for PCR and further analysis of phyla and genera in gut microbiomes of pigs. PCR products were sequenced in Illumina Sequencer at the Genomics Core facility at the Pennsylvania State University. PCR mixtures (50 μl) contained PCR buffer, 0.2 mM of each of dNTP, 0.4 mM each primer (Integrated DNA Technology, Coralville, IA), 2.5 U of FastStart high fidelity Taq polymerase, and 50 ng template DNA. PCR was performed using thermocycler (Eppendorf, Germany) programmed to perform 95 °C for 2 min, followed by 30 cycles of 95 °C for 30 sec, 60 °C for 30 sec, 72 °C for 30 sec followed by final extension at and 72 °C for 7 min. PCR products were visualized by agarose gel electrophoresis and DNA band excised, purified from the gels by Qiaquick columns (Qiagen, Valencia, CA) PCR products were sequenced on Illumina Sequencer at Genomics Core facility at the Pennsylvania State University. The samples were barcoded; taxonomic assignments of the sequenced 16S rRNA gene were made using the Ribosomal Database Project (RDP) Web tools (Roche Diagnostics, Indianapolis, IN).
Following sequencing, the barcodes were sorted, the 16S rRNA sequences were screened for quality. The sequences that did not match the primer sequences (=20-30) in the beginning and end of the reads were eliminated to minimize errors. The average total number of reads was 12,362,217, with an average number of 3,171,719 undetermined reads. RDP database was used for taxonomic grouping with a boot strap cut off of 80% for statistical analysis [20]. To determine the operational taxonomic unit (OTU) Mothur was used [21] with a definition at a similarity cutoff of 95%.
Microbial DNA qPCR array (QIAGEN, Carlsbad, CA) for detecting and relative profiling of 87 antibiotic resistance genes was used according to the manufacturer’s protocol. DNA isolated directly from fecal samples and DNA isolated after growing the fecal samples in TSB from pigs grown in conventional and organic dietary regimens were used for testing the presence of antibiotic resistance genes. The antibiotic resistance genes mPCR reactions were carried on StepOnePlus (Applied Biosystems) instrument and CT values of the two assays were averaged.
High-throughput sequencing technology of V3 region of 16S RNA was employed to determine the changes in the microbiota of swine in response to dietary antimicrobial supplements. The nucleotide sequences generated in a single run [22] was used for the metagenomics sequencing data analysis of the fecal samples of pigs grown in conventional and organic dietary conditions. The fecal bacterial population was also cultured in TSB media allowing the aerobic bacteria to grow for 12 hours and the metagenomics data for the direct fecal sample and the cultured samples for organic and conventional diets were compared. This experiment reflected the native microbial community of the gut and the changes in the composition through the enrichment process. The data depicted in Figure 1 shows that the phyla that are associated with microbiomes of pigs grown under conventional and organic diet, do not differ much. Presence of Bacteriodetes, Fermicutes and Proteobacteria were observed in both samples above 0.1% level. At the genus level, the gut microbiome in swine grown under conventional and organic diet regimens exhibited similar bacterial genera, such as Prevotella, Lactobacillus, Barnesiella, Proteus, Fecalibacteria, Escherichia coli and Shigella. There was little difference in the profiles when the fecal samples were cultured and enriched overnight between the two dietary regimens although some of the aerobic bacterial population grew under those conditions (Figure 2). In a recent extensive study from Europe, no differences in microbiota of pigs from conventional and organic farms were observed [23]. Similar observations were also made in earlier reports [24,25] where swine gut microbiota was shown to be comprised of mostly Bacteroidetes and Firmicutes. At the genus level, Prevotella, Lactobacillus, Treponema, Roseburia, and Streptococcus were commonly observed [24-27]. Looft et al. [28] demonstrated that Firmicutes to be most prevalent at the phylum level and Anaerobacter, Turicibacter, and Escherichia at the genus level in microbiota in swine ileum. Recently, Holman and Chenier (2014) [24] using Illumina sequencing of the V4 region of the 16S rRNA gene investigated the effect of sub-therapeutic levels of in-feed tylosin and chlortetracycline over a 19-week period The archaeal and bacterial diversity in pigs that were fed tylosin (11 to 44 mg/kg; 423 feed) did not change from controls. There were shifts observed in tylosin treated pigs in phyla and genus. While the genera Akkermansia, Coprococcus, and Streptococcus were enriched in tylosin-fed pigs, Bacteroidetes and the genera Succinivibrio and Anaerovibrio were reduced. However, in the presence of chlortetracycline (5.5 mg/kg) in feed, Firmicutes and Lactobacillus were altered [24].
The fecal samples from both groups were cultured overnight and their microbiotas were analyzed as described. There was practically no difference in the composition in samples that were cultured in enriched media versus those that were not, in both organic and conventional diet regimens. It could be because it is difficult to reproduce the gut microenvironment since many of these bacteria in the gut are anaerobes and have symbiotic relationship [29]. Only 1% of the bacteria are culturable [30]. Therefore, culturing of the samples exhibited slight shift in the gut microbiome towards the presence of bacteria at the genus level as some of the aerobic bacterial population grew under those conditions (Figure 2). There are reports that found different microbiota in nursing sows versus those that are post- weaning [24,31]. In our experiment, sampling was conducted from pigs that were post-weaning, that exhibited no substantial difference in the microbial profiles in organic and conventional production. The genera that were abundant in the heat map included Roseburia, Prevotella, Lactobacillus, Hallela and Barnesiella, in conventional diet regimen. However following enrichment abundance of Succinivibrio, Proteus, Parasutterella, Oscillibacter, Faecalibacterium, Escherichia/Shigella, Erysipellotrichaceae, Dorea, Coprococcus, Barnesiella, Allisonella and Acidaminococcus were observed. Fecal samples from pigs grown under organic regimen exhibited the presence of Roseburia, Prevotella, Lactobacillus, Hallela and Clostridium. Following enrichment, abundance of Sutterella, Roseburia, Prevotella, Lechnospiracea, Hallela, Faecalibacterium, Erysipelotrichaceae, Dorea, Coprococcus, Barnesiella, Allisonella and Acidaminococcus were seen. The percent of phyla, families and genus in the microbiomes are depicted in Supplemental Table 1.
Antimicrobial resistance genes for floroquinolone, aminoglycoside, beta-lactamase, macrolide, tetracycline and vancomycin were detected in fecal samples from pigs grown in both conventional and organic diets as shown in Table 1. While floroquinolone resistance genes were observed in fecal samples in pigs grown under both dietary condition, beta lactamase class A and class B resistance genes (CTX M-1, SHV, KPC) were more abundant in conventional dietary regimen than organic. Beta lactamase class C genes were less abundant as compared to beta lactamase class D in both organic and conventional samples. Some fluorquinolone genes were observed in organic and some in conventional dietary conditions. Genes for macrolide/lincosamide, multidrug efflux, tetracycline efflux, vancomycin resistance and beta lactum resistance were observed in both regimens. The antimicrobials tetracyclines, macrolides, lincosamides and bacitracin have been used in swine production and resistance to these antimicrobials have been also observed commonly [32,33]. Since tylosin, tetracyclines and lincomycins are used also in human medicine; microorganisms resistant to these antibiotics are considered as serious threat worldwide by World Health Organization. These antibiotics when used sub-therapeutically by swine industry may potentially lead to the emergence of antibiotic resistant bacteria.
From the experiments it is not clear whether raising pigs under organic conditions assists in reducing antimicrobial resistance of microorganisms in gut microflora. Similar results have been reported by Gerzova et al. (2015) [23] who also did not find much difference between the abundance of tested antibiotic resistance genes in microbiota originating from organic or conventionally housed pigs in various countries. In one study conducted in China [34], evaluated the effect of feeding lincomycin hydrochloride (500 mg/kg; 447 feed), chlortetracycline (100 mg/kg) feed, and amoxicillin (500 mg/kg; 448 feed) to sows before farrowing over a 12-day period. Using real-time PCR [34] showed greater numbers of aminoglycoside, lincomycin, macrolide, and tetracycline resistance genes following antibiotic treatment in swine gut. This could be because of administration of large concentrations of the antibiotics to sows.
Recently Agga et al. (2015) reported higher presence of tet(A) gene but not tet(B) in the feces of post-weaning pigs that were fed chlorotetracycline (550 mg/kg; 477 feed). Others [12,35-37] have reported that there are antimicrobial resistant determinants observed in guts even when the pigs were not exposed to antibiotics in modern production facilities. In particular, the tetracycline resistance genes tet(M), tet(O), tet(Q), and tet(X) [36-38] and the macrolide resistance genes erm(B), erm(F), and erm(G) [12,28,37] are frequently detected in pigs not directly exposed to antimicrobials.
In conclusion, this pilot study conducted on the composition of microbiomes from swine grown in conventional and organic diet regimen show small differences in the presence of microorganisms at the phyla level and at the genera level (Figure 1 and 2). Both the groups exhibited presence of antimicrobial resistance genes, more so in conventionally grown pigs, although the organic group did not get exposed to antibiotics. Larger studies with more samples may provide evidence of any difference in microbiomes of swine that are grown in organic and conventional farms.
The authors like to acknowledge the support they received for conducting the research from the Animal Health and Diagnostic Commission of the Pennsylvania Department of Agriculture, USA.
We would like to thank Aswathy Sabastian and Dr. Istvan Albert, Huck Institute, Penn State for helping us to analyze the sequences.

![]()

 |
| Figure 1: 16S Analysis of Abundant Phyla from Microbiome of Fecal Samples of Pigs |
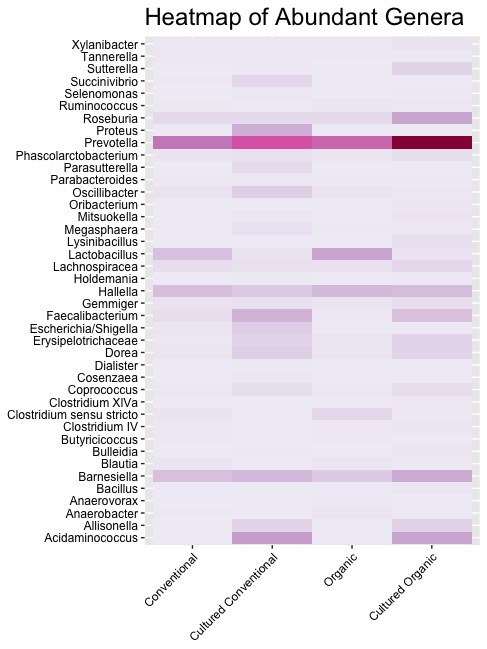 |
| Figure 2: Heat Map to Show the Abundance of Different Genera in the Gut Microbiomes |
Gene (NCBI Tax ID) |
Sensitivity |
Antibiotic Classification |
Organic |
Conventional |
CO |
CC |
|---|---|---|---|---|---|---|
AAC(6)-lb-cr |
100 |
Fluoroquinolone |
positive |
positive |
positive |
positive |
aacC2 |
30 |
Aminoglycoside |
negative |
positive |
negative |
positive |
aacC4 |
20 |
Aminoglycoside |
positive |
positive |
positive |
positive |
aadA1 |
200 |
Aminoglycoside |
positive |
positive |
positive |
positive |
aphA6 |
40 |
Aminoglycoside |
negative |
negative |
negative |
positive |
CTX-M-1 Group |
50 |
beta-lactamase, Class A |
negative |
negative |
negative |
positive |
KPC |
40 |
beta-lactamase, Class A |
negative |
positive |
positive |
negative |
Per-1 group |
30 |
beta-lactamase, Class A |
positive |
positive |
positive |
positive |
SHV |
200 |
beta-lactamase, Class A |
negative |
negative |
positive |
positive |
SHV (156D) |
100 |
beta-lactamase, Class A |
negative |
negative |
negative |
positive |
SHV (156G) |
50 |
beta-lactamase, Class A |
negative |
negative |
positive |
positive |
SHV (238G 240E) |
30 |
beta-lactamase, Class A |
positive |
negative |
positive |
positive |
SHV (238S 240E) |
50 |
beta-lactamase, Class A |
negative |
negative |
negative |
positive |
SHV (238S 240K) |
30 |
beta-lactamase, Class A |
positive |
positive |
positive |
negative |
IMP-5 group |
200 |
beta-lactamase, Class B |
negative |
negative |
negative |
positive |
VIM-13 |
20 |
beta-lactamase, Class B |
negative |
negative |
negative |
positive |
ACT-1 group |
100 |
beta-lactamase, Class C |
negative |
positive |
negative |
negative |
CFE-1 |
50 |
beta-lactamase, Class C |
negative |
negative |
positive |
negative |
DHA |
20 |
beta-lactamase, Class C |
negative |
negative |
negative |
positive |
FOX |
100 |
beta-lactamase, Class C |
positive |
negative |
negative |
negative |
LAT |
100 |
beta-lactamase, Class C |
positive |
positive |
positive |
positive |
MIR |
30 |
beta-lactamase, Class C |
negative |
negative |
negative |
positive |
OXA-10 group |
20 |
beta-lactamase, Class D |
negative |
negative |
positive |
negative |
OXA-18 |
30 |
beta-lactamase, Class D |
positive |
positive |
negative |
positive |
OXA-2 group |
40 |
beta-lactamase, Class D |
positive |
positive |
positive |
positive |
OXA-23 group |
50 |
beta-lactamase, Class D |
positive |
positive |
positive |
positive |
OXA-24 group |
20 |
beta-lactamase, Class D |
positive |
positive |
positive |
negative |
OXA-50 group |
20 |
beta-lactamase, Class D |
positive |
negative |
negative |
negative |
OXA-60 |
30 |
beta-lactamase, Class D |
negative |
negative |
negative |
positive |
QnrB-5 group |
40 |
Fluoroquinolone |
negative |
positive |
negative |
negative |
QnrB-8 group |
20 |
Fluoroquinolone |
negative |
negative |
negative |
positive |
QnrC |
30 |
Fluoroquinolone |
positive |
negative |
negative |
negative |
QnrD |
40 |
Fluoroquinolone |
positive |
positive |
positive |
positive |
QnrS |
40 |
Fluoroquinolone |
positive |
positive |
positive |
positive |
ermA |
100 |
Macrolide/Lincosamide |
positive |
positive |
positive |
positive |
ermB |
20 |
Macrolide/Lincosamide |
positive |
positive |
positive |
positive |
ermC |
100 |
Macrolide/Lincosamide |
positive |
negative |
positive |
positive |
mefA |
100 |
Macrolide/Lincosamide |
positive |
positive |
positive |
positive |
msrA |
100 |
Macrolide/Lincosamide |
positive |
negative |
negative |
negative |
oprj |
50 |
Multidrug efflux |
negative |
positive |
negative |
negative |
tetA |
40 |
Tetracycline efflux |
positive |
positive |
positive |
positive |
tetB |
30 |
Tetracycline efflux |
positive |
positive |
positive |
positive |
vanB |
100 |
Vancomycin resistance |
positive |
positive |
negative |
positive |
mecA |
40 |
Beta-lactum resistance |
positive |
negative |
positive |
negative |
CO: Cultured organic; CC: Cultured conventional |
||||||




































