Open Access
Case report
Max Screen >>
ISSN: 2455-7617
Copyright: © 2016 Simman R. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Warfarin is the most frequently prescribed oral anticoagulant in the United States. Its indications range from treatment of deep venous thrombosis and pulmonary emboli to the prophylaxis and treatment of thromboembolic conditions associated with atrial fibrillation and cardiac valve replacement. While bleeding is the most commonly encountered complication warfarin induced skin necrosis (WISN) can be a rare complication. Extensive skin and deeper tissue necrosis may be encountered requiring multiple surgical debridement and reconstructive procedures along with the medical management.
With this disease tissue necrosis of the lower extremities involved not only the skin but deeper layers including subcutaneous tissue, tendons and bones suggesting a more accurate terminology to depict the condition would be warfarin induced tissue necrosis (WITN).
Keywords: Warfarin; Tissue necrosis
Warfarin is a commonly prescribed medication worldwide, with indications for treating acute deep vein thrombosis, pulmonary emboli and prophylactically to prevent a thrombotic event associated with atrial fibrillation and cardiac valve replacement. [1] while bleeding is the most common and serious side-effect of this medication [1], WITN is a rare complication encountered. Estimated to effect 0.01 to 0.1% of all patients treated with warfarin and it cogeners [2-6], only about 300 cases have ever been cited in the medical literature [6]. This condition often results in extensive skin and deeper tissue necrosis requiring plastic and reconstructive surgeon intervention. It is necessary for clinicians to be familiar with the identification, differentiation, treatment and management of this condition. We propose to change the name of the condition to Warfarin-Induced Tissue Necrosis since tissue involved may go deeper than just the skin to include subcutaneous tissue, tendons and bones.
A 59-year old Caucasian male, with a past medical of chronic systolic congestive heart failure, presented to his primary care physician for worsening lower extremity edema. The patient was found to be diagnosed with new-onset atrial fibrillation and acute on chronic decompensated systolic heart failure. Treatment was initiated for both conditions and included starting the patient on Warfarin for oral anticoagulation to prevent thromboembolism. He was started on daily Warfarin at that time. He subsequently presented to the emergency department 14 days after starting warfarin with worsening swelling of both lower extremities and associated tenderness. His other complaints included epistaxis, bloody diarrhea and a localized, hemorrhagic area with blisters on the left lower extremity since starting warfarin. At that time he was also found to be in atrial fibrillation with rapid ventricular response. Shortly after, he developed extensive skin necrosis involving his legs and feet bilaterally. Dry gangrene was also noted on his toes (Figure 1, 2 and 3).
Scattered ecchymosis was noted on his left upper extremity. Work up in the emergency department showed acute deep vein thrombosis seen on venous ultrasound with partial thrombus within the left common femoral vein to the distal femoral vein. Arterial ultrasound showed partial occlusion of the peroneal artery. Initial coagulation profile showed PT greater than 270, APTT was 174 and INR was undetectable. Further initial testing at the time of admission a 1:1 mixing study was performed that did not correct and was found to have a low antithrombin activity. There was no known family history of hypercoagulable disorders and no previous exposure to heparin products. Other comorbidities treated during his hospital stay included acute renal failure requiring dialysis, a bleeding duodenal ulcer requiring transfusion of fresh frozen plasma and packed red blood cells, acute respiratory failure from a pulmonary embolism requiring intubation, as well as pseudomonas bacteremia. Initially Coumadin was held and coagulopathy was reversed, low dose heparin was used for symptomatic thrombotic episodes secondary to consumptive coagulopathy. Initial laboratory work up included: normal factor V Leiden and normal anti-cardiolipen IgG and IgM antibodies. Heparin induced thrombocytopenia assay was non-reactive. Factor VIII was elevated, factor II, VII, IX, X and XII were low.
Disseminated intravascular coagulation (DIC) panel showed elevated fibrinogen, D-dimer, activated partial thromboplastin time (aPTT) and prothrombin time (PT) and international normalized ratio (INR 3.5). Lupus anticoagulant was negative. He was continued on a therapeutic dose of heparin and an inferior vena cava filter was placed. Once stabilized in the acute care setting, the patient was transferred to a long term acute care hospital on low dose warfarin and heparin for bridge therapy till his INR was therapeutic.
The necrotic skin areas were initially treated with daily dressing change using Xeroform gauze and kerlex wraps. After the acute phase of his hospital stay resolved, he was taken to the operating room for extensive sharp debridement of the necrotic tissue. The right lateral leg had necrosis extending down to the level of the periosteum of the fibula. The left dorsum of the foot had full thickness necrosis, involving skin, deep subcutaneous tissue, extensor tendons and tarsal and metatarsal bones. After about a month, a second debridement surgery of the right lateral leg, left foot dorsum, and left heel was completed. At this point, many of the wound beds had initiated the healing process and extensive granulation covered the remainder of the wounds. A third debridement of the same areas, plus an amputation of the 3rd toe of the left foot, occurred about 2 months after the initial debridement. Figures 4 and 5, show a preoperative view of the left foot. At this time, a biosynthetic collagen-based extracellular matrix, derived from porcine small intestine submucosa, was applied to areas with exposed tendons and bone, in order to promote granulation tissue and prepare the wound bed for autologous skin grafting. Three weeks later, he required another debridement of the left foot dorsum, a meshed split thickness skin graft was taken from the right lateral thigh and was applied to all area of the lower extremities that had good granulation tissue including the right lateral leg and the dorsum of left foot (Figure 6). Tarsal bone osteomyelitis was diagnosed with bone cultures growing pseudomonas bacteria was treated with IV antibiotics based on culture sensitivities. Through repeat debridement, the usage of advance wound care technology, and the application of skin grafts, the patients' lower extremities heel and amputation of the extensively damaged lower legs and feet was avoided (Figures 7, 8, 9, 10, 11 and 12).
Warfarin, which was been around since the early 1940's, is the most frequently prescribed oral anticoagulant in the United States [7]. The condition of WITN was thought to be first described by Flood et al in 1943 [8] and then again by Verhagen et al in 1954 [9]. The first case series in the United States was reported by Kipen [10] in 1961 and since then, three major literature reviews have been published describing a compellation of the characteristics associated with WITN [7,11-13]. Classically, WITN presents in middle aged, obese women that are being treated for a deep vein thrombosis or a pulmonary embolism [6,11]. However, since the literature on this condition is limited to small case reports, the true demographical factors representing WITN is unknown. The first skin symptoms appear to present with a range of 1-10 days with the vast majority presenting in the interval of 3-6 days [5,6,9,11]. There have also been numerous cases presenting outside this window [5,14], so keeping this condition as a differential diagnosis of necrotic skin lesions is prudent. WITN has a propensity for areas with large amounts of subcutaneous fat, such as the breast, buttock, and thigh. However, WITN lesions have been identified in most area and can be unpredictable [6].
Pain, parasthesia, and sensation of pressure are often non-descript symptoms prior to skin manifestations [6,7]. Associated skin changes typically begin with an evanescent flush, followed by a petechial rash and a sharply demarcated ecchymosis with an erythematous rim [7]. Hemorrhagic bullae and eschar development is the terminal events of irreversible full-thickness necrosis [6].
Keeping a broad differential is of paramount importance when approaching a suspected case of WITN. WITN can mimic calciphylaxis, micro-embolization (septic emboli, cholesterol emboli), heparin-induced skin necrosis as a result of the thrombocytopenia and thrombosis syndrome, DIC, purpura fulminans, necrotizing fasciitis, cryoglobulinemia, inflammatory breast cancer, decubitus ulcers, and snake venom induced skin necrosis [5]. A detailed history in conjunction with timing, progression, and characteristics of the skin necrosis will help to differentiate WITN from the other above causes. In addition, laboratory data and biopsy information may help to solidify the diagnosis going forward.
Currently, several theories exist that try to explain why WITN happens. However, the exact mechanism of the pathology that is happening remains to be elucidated. There are several associated hereditary conditions that seem to exacerbate and may even promote the development of WITN, include protein C deficiency, protein S deficiency, antithrombin III deficiency, factor V Leiden mutation, and antiphospholipid antibody syndrome. [3,5,6,15-17] It was proposed that warfarin had a direct toxic effect (toxic vasculitis) at the junction of the precapillary and arterial capillary of the dermovascular loop [11]. The drug-damaged capillaries dilate and rupture causing a petechial rash. Veins distal to the injured capillaries thrombose creating stasis of blood and ultimately tissue necrosis. Consequently, arteries in the skin and subcutaneous tissue are spared, whereas capillaries, venules, and, occasionally subcutaneous veins are occluded. Following warfarin administration, plasmaprotein levels are believed to drop precipitously before the anticoagulant effect commences. An imbalance between procoagulation and anticoagulation occurs with the creation of a hypercoagulable state.
Warfarin inhibits vitamin K dependent clotting factors which include factors II, VII, IX and X [19,20]. While warfarin inhibits procoagulants it also has a potentially thrombotic effect by impairing synthesis of Proteins C and S, two inhibitors of coagulation that have the shortest half-life. The full effect on warfarin is seen when all vitamin dependent factors have decreased, this takes 36-72 hours with a steady state occurring at approximately seven days. For this reason, in acute thrombotic events, there is a required overlap of low molecular weight heparin and warfarin for 4-5 days to avoid worsening or new thrombosis [21]. Additionally, starting warfarin at a dose over 5 has a potential complication of transient procoagulant state from precipitous decrease in protein C within the first 36 hours. Additionally factor VII decreases which may increase the INR without fully decreasing factor II which is when there is a full anticoagulant state.
Another complication seen with higher doses of warfarin within the first few days of initiation is skin necrosis. Necrosis has been shown to be mediated by the decrease in protein C. Approximately 1/3 of the patients with warfarin induced skin necrosis were diagnosed with protein c deficiency. Despite this finding, it is still a rarely seen in patients with protein C deficiency [22]. Other case reports show an association with protein S deficiency and factor V leiden.
This inequity generates thrombotic occlusions of the microvasculature with resulting necrosis.
Within the clotting cascade, protein C works together with protein S. With activated protein S, protein C inactivates factors V and
VIII a resulting in decreased thrombin, prothrombin and increases fibrinolysis [23]. Acquired states of protein S deficiency can occur in pregnancy and oral contraceptive use, as well as DIC and acute thromboembolic events [24].
When testing for suspected hypercoagulable disorders it is important to consider timing of testing. Checking for deficiencies in antithrombin, protein C and S should be done while on heparin or after 2 weeks of discontinuing warfarin. Testing while on warfarin testing may result in falsely low or functionally low levels, and if levels are normal they need to be rechecked at a later date to confirm values [25,26].
DIC is characterized by forming both thrombus and hemorrhage and can occur as an acute or chronic process. This needs to be considered in the differential diagnosis as the presentation may be similar. Common manifestations of the disease are ecchymoses, petechiae, active bleeding, venous and arterial thrombus [27]. Purpura fulminans is also a rare disease that is associated with DIC along with tissue thrombosis and hemorrhagic skin necrosis and Inherited protein C deficiency. In DIC, testing shows prolonged PT and aPTT, elevated INR, hypofinbrinogenemia, elevated D-dimer, thrombocytopenia and microangiopathyc hemolytic anemia.
Despite proposed etiologies and association of an underlying coagulopathy, the majority of patients with WITN do not have an identifiable inherited hypercoagulable state, and whether a causal relationship exists in those who do is uncertain. Also noteworthy is that WITN rarely occurs in ambulatory settings, such as in new-onset atrial fibrillation patients like the case presented here. Perhaps an acute procoagulant process, such as an active thrombus, may be necessary along with other unknown mechanisms. Another important aspect is that WITN is a complication of the drug therapy rather than the result of simple over-anticoagulation.
The current treatment approach to patient with WITN has two parts. First, stop the offending agent (warfarin), place the patient on an alternative anticoagulant, and replenish the patients' supply of vitamin K and coagulation factors inhibited by warfarin (Factors II, VII, IX, X, Protein C, and Protein S) [19,20]. Part two is the surgical approach which may be needed to aid in optimizing wound healing including the removal of the necrotic tissue and coverage of the resulting defect with skin grafts or flaps.
Warfarin induced tissue necrosis, while rare, should be considered in a patient with cutaneous lesions. While it is important to keep a broad differential, early diagnosis and treatment with a team approach is necessary to improve the outcome. As demonstrated in this case the tissue damage was deeper than just the skin and it involved the extensor tendons and the underlying bone of the foot. As a result, we propose to change the name to WITN.

![]()

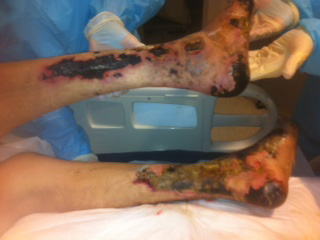 |
| Figure 1: Posterior view of bilateral legs and ankles tissue necrosis |
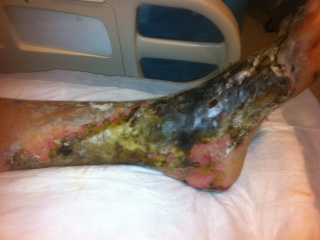 |
| Figure 2: Closer view of left foot and ankle with extensive tissue necrosis |
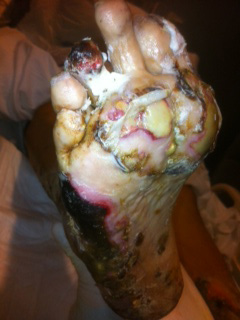 |
| Figure 3: Plantar right foot with tissue necrosis |
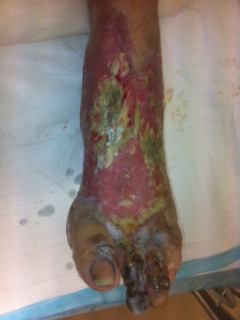 |
| Figure 4: Left foot with second toe gangrene, third toe partial necrosis, proximal first, second and forth metatarsal bones necrosis and pealing soft tissue eschar |
 |
| Figure 5: Closer view of dorsal left foot with necrotic proximal metatarsal and navicular bones |
 |
| Figure 6: Left foot after skin grafting and Integra application over after debrided necrotic navicular and metatarsal necrotic bones |
 |
| Figure 7: Healed heels and posterior legs wounds |
 |
| Figure 8: Healing right plantar foot wounds |
 |
| Figure 9: Healing right plantar forefoot wounds |
 |
| Figure 10: Healed plantar right foot wounds |
 |
| Figure 11: Healed second toe amputation and partial third toe amputation and skin graft at the foot dorsum |
 |
| Figure 12: Healed left foot graft and amputation sites |




































