Open Access
review article
Max Screen >>
ISSN: 2576-7690
Copyright: © 2015 Ahmad Y. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
In the post genomic era when several proteomes are on the verge of completion, promising field of protein based diagnostic techniques is emerging. Although protein detection have been used for a long time in clinical diagnostic test, yet high throughput proteomics approaches along with systems biology could be a step forward, towards the development of next generation diagnostic tools and pave a way for personalized medicine. Moreover, the integration between proteomics, genomics, transcriptomics, bioinformatics and biostatistics has allowed for the development of an important instrument for translational aspects of modern molecular medicine. This review familiarizes young researchers with historical aspects of diagnostic proteomics, its current applications and a hypothetical view on prospective techniques with potentials for diagnostic capacities.
Keywords: Proteomics; 2DE; MALDI-TOF; Diagnosis
Proteome was coined by Marc Wilkins, which he used for the entire complement of protein expressed by a genome, cell, tissue or organism. Proteomics is the study and characterization of complete set of proteins that are present in cell, organ, or organism at a given time [1]. Unlike genomes proteomes have both temporal and spatial variations which escalate the complexity of its study. However, this variation is important for clinical diagnosis of various pathological conditions, as a minute change in cellular or subcellular metabolism may alter its proteome [2]. Last decade has witnessed tremendous increase in the whole genome sequences. As per the recent data from genome online database (GOLD), there are 9191 eukaryotic genomes sequenced so far, while there are 38967 genomes available within bacterial domain [3]. This exponential rise is however not concurrent with proteome maps, perhaps due to lack of advent in proteome analysis technologies and also several challenges due to dynamic nature of proteomes. The dynamics of proteome is further complicated by post translational modifications that regulate the protein activity that creates a difference between proteome and functional proteome, one of the emerging fields in proteomics.
Several metabolic diseases and genetic abnormalities are known to impair the cellular transcription and translation status and thereby creating a variation in proteome [4], in fact subcellular proteomes such as mitochondrial and nuclear proteomes are also affected in limited condition [5]. These alterations in proteome are an opportunity for development of diagnosis of molecular signatures for several diseases and lead to the development of a new domain called 'clinical proteomics'. Classically, clinical proteomics is defined as clinical and analytical validation and implementation of novel therapy or diagnosis related markers that originate from preclinical studies designed to identify leads in analogy to drug screening studies [6]. Serological examination of immunoglobulin's or antigens are common tools in most of the diagnostic methods, however assessment of proteome or part of proteome is advantageous as the dynamics of proteomic changes is much rapid in contrast to immunological responses [6]. Proteomics tools have evolved during the last decade, several new techniques have emerged that can allow separation and relative quantification of proteins in small time and at low cost, however several limitations prevent them from reaching the bedside [7]. Below are some of the common proteomics techniques subdivided into gel-based and gel-free, which are based on the initial separation step followed by visualization, analysis and identification steps that are either in use or have prospective use in clinical proteomics. The separative step aims to resolve complex protein mixtures in order to separate each protein species contained inside the sample. The separation process easily permits the subsequent quantification and identification steps.
One dimensional gel electrophoresis is based on electrophoretic mobility of proteins on an inert support such as polyacrylamide and agarose that results in separation of proteins on the basis of their molecular weight. Any shift or missing bands might represent alteration in the proteome status. Several variants of the gel electrophoresis have been developed, Native-polyacrylamide gel electrophoresis (PAGE) involves the use of proteins in native state and therefore use of denaturing conditions such as mercaptoethanol, dithiothreitol, or sample boiling is eliminated. This technique is useful for determination of biological activity of proteins in gels.
A more specific and sensitive set of proteomics technique include immunoaffinity based techniques. In these techniques antibodies against a specific protein of interest (may be a biomarker or pathogen- antigen) are specifically targeted and identified. One of the most popular techniques in use is ELISA (Enzyme linked immunosorbent assay). This technique has several variants such as direct, sandwich and competitive ELSA. Direct ELISA involves the detection of antigen in the plasma by probing it with antibodies and then secondary antibodies conjugated with an enzyme are used to develop specific colour using chromogenic substrate. Indirect ELISA and sandwich ELISA involve the detection of antibodies present in the serum of patient [8]. Whereas in competitive ELISA the antigen titer is detected by using a competitively labeled antigen that primarily enhances the sensitivity of the assay. However, this format is less often used in diagnosis due to complex assay procedures [9]. Another set of immunological tools called immunoprecipitation involve the detection of soluble antigens, especially those of viruses and small pathogenic bacteria. In this technique antibodies or antigens can be added to sample to observe a visible precipitation. Preferably the precipitation is observed on an inert gel support such as agarose and known as gel precipitation. Application of electric current to enhance the mobility of antigen or antibody on gels is often used to reduce time to carry out experiment and this is known as immunoelectrophoresis.
Two dimensional electrophoresis has preceded and accompanied the birth of proteomics. Established in the early '70s, thanks to O'Farrel, still remains the technique of choice for this kind of study although lately interest has focused on the development of gel-free techniques. The 2DE is reproducible, robust and able to best resolve a complex protein mixture according to their isoelectric point (pI) and their molecular weight (MW). The polyacrylamide gels, where the protein species are resolved, represent the "core" of the proteomic analysis. Its structure consents to physically match two different samples. Thanks to this, two different protein mixtures are compared to each other in both quantitative and qualitative points of view. Another advantage of the 2DE method is the study of post-translational modifications that determined an alteration of the pI and MW inducing a positional shift in the 2D gel. This kind of modification is represented by phosphorylation, glycosylation; glutathionylation or more neglected modification such as protein cleavage. In conclusion, the 2DE for separative step is decisive for the next selection of interesting spots for the analysis. Although widely used, 2DE presents some limitations: the reduced dynamic range, for instance, allow the visualization of the under-represented proteins limiting the global approach of the proteomic method. In addition, 2D gels also rarely display hydrophobic proteins and only highly abundant proteins are currently visualized. Low abundance proteins of physiological relevance, such as regulators or signaling proteins are difficult to detect. Moreover, basic or very basic proteins are rather difficult to focus. In addition to these technical problems, 2DE is a "time-consuming" method that makes it possible to carry out a comparison of a low number of analytical and biological replicates.
In support of the various problems encountered with classical 2DE on gel-to-gel variations and time-consuming questions, DIGE has been developed which substantially reduces variability by sample labeling with different fluorescent dyes (Cy2, Cy3, and Cy5). In the same gel it is possible to resolve control and treated samples labeled independently with a fluorescent dye such as Cy3 or Cy5. Cy2 allows labeling an internal standard, a mixture containing equal amounts of each experimental sample taken into consideration. Two samples and the internal standard are mixed together and resolved in the same gel. Densitometric scanning at different wavelengths, characteristic for each dye, permit to obtain three images from only one gel, two from samples and one from internal standard. This procedure allows a very accurate and fast computer analysis reducing errors due to the distortion of the experimental gels. The internal standard represents the average of the analyzed samples reporting every protein species. Its use allows an accurate statistical spot quantification as well as an increase in matching gel reliability to distinguish the experimental from biological variations in the samples. The classical or DIGE gel production needs an image analysis step by dedicated software such as Image Master 2D Platinum (GE Healthcare, Uppsala, Sweden) for classical gels and De Cyder (DeCyder Differential Analysis software, GE Healthcare) with regard to the DIGE gels. The usefulness of DIGE is amplified multifold by the use of mass spectrometry.
Edman degradation, a method for proteomic sequence also emerged as an important tool in determination of proteins sequences and growth of the proteome databank pools of several organisms. However, the complex nature of technique and advent of bioinformatics tools to obtain protein sequence from genomic data downgraded the importance of protein sequencing. Currently, mass spectrometric techniques have also matured as a tool for determination for protein sequences by using MSn.
Mass spectrometry (MS) is based on the identification of a molecule on the basis of small molecular fragments created by high energy laser and detected on the basis of their different mass to charge (m/z) ratios. MS was classically a tool for chemists for the identification of relatively smaller molecules however the usefulness of mass spectrometry was focused in early 90s when Tanaka et al, demonstrated the identification of proteins using matrix assisted laser dissociation spectrometry (MADLI). Perhaps, this technique should be considered as a quantum leap for the rapid growth of proteomics in the last decade [10]. In MALDI ToF procedure, the protein spot of interest resolved by 2DE is previously subjected to hydrolytic cleavage by trypsin. This enzyme cleaves the peptidic chain at arginine and lysine levels. The digested mixture of peptides is then, spotted onto the target plate, along with suitable matrix (Saturated solution of α-cyano-4-hydroxycinnamic acid). MALDI is a mild ionization technique allowing the analysis of biomolecules which tend to be fragile and fragment when ionized by more conventional ionization methods. In the first part of MALDI ToF analysis, aromatic groups of the matrix absorb the laser energy ionizing its acidic group. This process consents to transfer a proton to the peptide. The desorption of the sample is achieved using vacuum in the flight tube. This process consents to obtain charged and in gaseous phase peptides that fly in the flight tube only depending on electromagnetic potential difference where every peptide is characterized by the same kinetic energy. What distinguishes the time of flight of each peptide is the time that the peptide employs to reach the detector starting from the target plate, will be its m/z ratio. Every peptide assumes a single charge (z = +1) hence, the mass will characterize each aminoacidic chain and then the time of flight. According to this, the smaller peptides will reach the detector before the bigger ones. The time of flight employed will be recorded and reported on a spectrogram, a graph showing the values of the m/z ratios on the x-axis and the intensity of each peak on the y-axis (each ion with the same m/z ratio). All the m/z values determine the peptide mass fingerprinting (PMF) of the protein, useful in comparing the experimental masses obtained from MALDI ToF, with the theoretical masses in specific databases available online on Swiss Prot (https://www. expasy.org/sprot/) and NCBInr (www.ncbi.nlm.nih.gov/protein). Mascot Search (www.matrixscience.com) is a research program, similar to Profound (https://prowl.rockefeller.edu/profound_bin/WebProFound.exe) able to perform the comparison between experimental and theoretical masses to identify the protein. The degree of identification accuracy is estimated by score value and sequence coverage. MALDI ToF technology is extremely versatile in proteomic analysis thanks to its capacity to generate monocharged ions and to its high sensitivity. The MALDI ToF can also be applied to the protein modification such as post-translational modification (phosphorylation, glycosylation etc) and protein interactions (protein-ligand or protein-metal ions). Later other variants of mass spectrometers based on electrospray ionization (ESI) and quadrupole detectors increased the sensitivity of protein identification. In contrast to the MALDI ionization, which leads to the mono-charged ion formation, ESI ionization leads to multi charged ion formation. The HPLC and ESI-IT conjugation allows increasing the spectrometer sensitivity because it is very dependent on sample entrance flow. Nanoliters/minute flow allows obtaining high sensitivity performance. Pros and Cons of the present and emerging proteomic techniques have been summarized in Table 1.
Above mentioned techniques are generally regarded as matured techniques, however several new techniques are progressively expanding and creating new dimensions for the clinical proteomics. The cost ineffectiveness of these techniques prevents them from reaching the diagnostic labs in developing and underdeveloped regions of the globe, despite of their tremendous capabilities. Perhaps, a revolution analogous to genomic sequencing techniques is awaited for the rapid capture of clinical diagnostic market that would reduce the cost of these techniques by several folds. Another limitation of proteomic diagnostic techniques is the unavailability of reliable information on dynamics of cellular or systemic proteomes [11]. Although the results are accumulating on proteome profiles of several pathogenic and multifactorial disorders, there is a strong need of integration of these data and development of in silico simulation techniques for proteome dynamics.
Clinical applications of proteomics tools begun much before the term' proteomics' was coined several techniques such as agarose gel electrophoresis for proteins, moving boundary electrophoresis and zone electrophoresis were popular in late 80s. Several studies on Hemoglobinopathies suggest the use of electrophoresis based diagnosis [12]. Sickle cell anemia and thalassemia traits were being detected with the help of several variants of gel electrophoresis [12,13]. In sickle cell anemia, mutation Q6V leads to the formation of structurally abnormal hemoglobin with diminished oxygen carrying capacity [14]. This serious condition is inherited in autosomal recessive fashion. Scientific literature from mid-80s to mid-90s reveals three common proteomics techniques in diseases diagnosis cellulose acetate/citrate agarose gel electrophoresis, capillary electrophoresis and isoelectric focusing Figure 1. Cellulose acetate/citrate agarose gel electrophoresis involves separating negatively charged proteins on an alkaline pH (8.2-8.6), due to variability in amino acid composition the charge of two molecules also varies and therefore two proteins with similar weight can also be resolved at different locations, this needed to be confirmed with running the gel in acidic conditions (pH 6.0- 6.2) in which samples are not being resolved on alkaline gel. In this method citrate was added to agar to make the surface acidic and then the charge of amino acids varied according to their respective isoelectric points. Often, this electrophoresis was applied to identification of Hemoglobinopathies [13]. Figure 2 illustrates the principle of cellulose acetate/citrate agar gel electrophoresis to resolve hemoglobin variants. Further additions were made to improve the specificity in diagnosis by integration of electrophoresis with immunological molecules popularly known as immunoelectrophoresis.
Radioimmunoassay is highly sensitive technique emerged in late 1970s. Hutchinson et al developed a simplified radioimmunoassay for diagnostic serology [15]. Later this technique was constantly used for the diagnosis of malaria [16], congenital adrenal hyperplasia [17] endocrine tumors of pancreas [18]. Lately, emergence of fluorescent immunocapture techniques with parallel sensitivity and safety superposed the radioimmunoassays.
Turbidometry has been widely used since the early 1950s as a method of quantitative analysis. This technique involves, the reduction of light intensity caused by interaction of light beam with suspension of particles, and further determined using spectrophotometry. During the early advent of this technique several diseases were diagnosed using this method. Detection of lipase activity of enzyme [19] and C reactive protein [20] is serum, diagnosis of pulmonary hypertension [21]. Nevertheless, conjugation of immunological molecules had improved scope and sensitivity of assay keeping them viable in current clinical diagnosis scenario.
Capillary electrophoresis (CE) equipped with a diode-array detector have been used to determine diagnostic protein biomarkers occurring in urine of patients with various diseases [22-24]. Identification of abnormal proteins was based on relative migration times and characteristic diode-array spectra (Figure 3). The CE-method readily diagnosed alkaptonuria, neuroblastoma and liver failure due to galactosemia. The simple and automated CE method appeared to be suitable for implementation as a screening test in analytical systems aimed at diagnosis of metabolic disorders, ulcerative colitis, Crohn's disease, Scrapie and thalassemia [22-24]. A summary of key techniques established in early 90s is described in Table 2.
Immunological techniques especially those based on selected antibodies for the detection of plethora of blood proteins identified as biomarkers are the most dominant diagnostic tools in use at present. ELISA is a diagnosis of choice by several clinicians primarily due to high sensitivity, specificity and ease of performing. Although several variants of ELISA have been commercialized by now yet the focus of detection remains associated with pathogenic diseases via direct ELISA where antigen is determined using specific antibodies [9]. Nevertheless, nonpathogenic diseases especially those involving inflammation and autoimmune disorders can also be detected with the recent expansion of the ELISA strategies [25]. At present clinically approved ELISA kits are available for the diagnosis of various allergies, anemia, autoimmunity, fertility, food intolerance, infectious diseases and inflammation (www. omegadiagnostics.com/products.). Among infectious diseases several viral diseases such as HIV infection, Lyme diseases and recently emerging Ebola virus are also diagnosed with the help of ELISA.
Electrophoretic techniques which were used in late 80s are now conjugated with immunological molecules and highly specific and sensitive technique immunoelectrophoresis is preferred over conventional electrophoretic methods. Several autoimmune diseases, enteropathies and waldenstrom's macroglobulinemia have been routinely diagnosed using immunoelectrophoresis [26].
Early 2000s, witnessed a further growth in clinical proteomics and older laboratory techniques such as mass spectrometry (MALDI), HPLC, immunoblotting matured over a period of time, bringing them close to other diagnostic tools. Early in the present decade 2D gel electrophoresis and MALDI based mass spectrometry has flourished and number of publications increased rapidly pertaining to their application in clinical reference [10]. These techniques brought a paradigm shift in the diagnosis and extended the usage to diagnosis of pathological diseases and multifactorial disorders, against the predominant diagnosis of Hemoglobinopathies in the 90s. Table 3 summarizes examples of diseases that have been successfully diagnosed with existing techniques. Two dimensional gel electrophoresis is a time consuming and complex technique to be carried to clinics, however this has proven to be an important tool research laboratories to narrow down hundreds of proteins to one or two significant proteins which could be called as 'biomarker' and therefore providing a molecule to clinicians which could be a gold standard for diagnosis of disease. Several biomarkers for multifactorial diseases such as diabetes, cancer, cardiovascular diseases have been reported till date. Nevertheless, prominent proteins from pathogens observed in host bodies could also be resolved using 2DE and identified as biomarker for pathogenic diseases, Molecules such asPro2PSA for identification of benign cancer, ROMA for prediction of malignancy of ovarian cancer, CA12-3 for disease progression of breast cancer. Current statuses of FDA approved biomarkers have been reviewed by Anna K Füzéry et al [27]. As discussed being a cumbersome and time consuming technique 2DE could not reach the clinics, however, Mass spectrometry, especially MALDI-TOF have successfully made its way to pathology labs in the present decade. Several diseases like cancer, Alzheimer's disease, Amyloid lateral sclerosis, renal amyloidosis, and Severe combined immunodeficiency can now be diagnosed using mass spectrometry and its variants [28-34].
Much advanced versions of mass spectrometry such as electron-spray ionization (ESI), liquid chromatography coupled mass spectrometry (LC-MS) have gained recent attention due to higher sensitivity and simplicity in the sample preparation.
Selected reaction monitoring (SRM) is recently emerged technique as an advancement of targeted proteomics. SRM has been employed for the consistent detection and accurate quantification of specific, predetermined sets of proteins in a complex background [35,36]. Technically, it is based on triple quadrupole (QQQ) mass spectrometers to selectively isolate precursor ions corresponding to the mass of the targeted peptides and to selectively monitor peptide-specific fragment ions. Suitable sets of precursor and fragment ion masses for a given peptide, called SRM transitions, constitute definitive mass spectrometry (MS) assays that identify a peptide and, by inference, the corresponding protein in proteome digests. SRM has high sensitivity (lowattomolar) and a broad dynamic range (up to five orders of magnitude), and it is quantitative. Key advantage of the SRM technique is its reproducibility and high sensitivity. These advantages advocate the prospective use of this technology in clinical diagnosis. At present the per sample cost of analysis is quite high and therefore beyond the reach of clinics in developing countries.
Advent of new technologies has brought with it the rapid growth in data accumulation especially discovery of new biomarkers and novel proteins differentially expressed in time and space. These data are certainly an addition to the current knowledge of diagnostic methods which will make the proteomic diagnosis much more sensitive, reliable and cost effective. Several new techniques are still in incubation and beings foreseen as future proteomic diagnostic tools. Surface enhanced laser desorption ionization (SELDI) is based on the adsorption of charged peptide fragments on a metal wedge to enhance the sensitivity for protein detection using mass spectrometry [37]. The mass spectrometric data that has been accumulated over a period of time is now being used to train the machines (alternatively called as artificial intelligence) so that mass fingerprint recognition could be made easy, these techniques use several computation tools and therefore enable proteome pattern recognition [38]. Conventional pI strips which were commercialized in year 2000 are now advancing towards much compact and high resolution formats called zoom gels [39]. Three dimensional gel electrophoresis is also being seeked as a better option for the improvement of resolution and reduction of protein separation time [40]. Parallel to microarrays, in which one can analyze experimental profile of thousands of m-RNAs, protein arrays have also emerged. However, the array formats are restricted to only part of proteomes and the operational cost is still higher. An improved version of protein arrays is a reverse phase protein array which is a key technique to study cellular circuitry and profiling of fundamental functional state of protein pathways [10]. Quantitative proteomics which has emerged as a new arm of proteomics is also expected to be a very promising technology as it would allow the absolute quantification of proteins in a given sample. The major label based quantitative proteomic techniques are under incubation namely, ICAT (Isotopic coded affinity tags), SILAC (Stable isotopic labeling with am and iTRAQ (Isobaric tags for relative and absolute quantification) [41]. In an ICAT experiment, protein samples are first labeled with either light or heavy ICAT reagents on cysteine thiols. The mixtures of labeled proteins are then digested by trypsin and separated through a multistep chromatographic separation procedure. Peptides are identified with tandem MS and the relative quantifications of peptides are inferred from the integrated LC peak areas of the heavy and light versions of the ICAT-labeled peptides [42]. Compared with the ICAT, a popular in vitro labeling, SILAC has emerged as a valuable in vivo proteomic technique which requires no chemical manipulation, and there is very little chemical difference between the isotopically labeled amino acid and its naturally occurring counterpart [43]. iTRAQ is another technique to perform absolute or relative quantification of protein in gel free system. This technology uses an NHS ester derivative to modify primary amino groups by linking a mass balance group (carbonyl group) and a reporter group (based on N-methylpiperazine) to proteolytic peptides via the formation of an amide bond [44]. Multidimensional Protein Identification Technology (MudPIT) is yet another emerging proteomics technique. In the MudPIT approach, protein samples are subjected to sequence specific enzymatic digestion, usually with trypsin and endoproteinase lysC, and the resultant peptide mixtures are separated by strong cation exchange (SCX) and reversed phase (RP) high performance liquid chromatography (HPLC) which are then identified using mass spectrometry [45]. This technique is also useful for the determination of protein-protein interactions [46]. Table 4 summarizes the emerging techniques with their possible applications and advantages [45].
Box 1: Effect of preanalytical techniques on protein
Preanalytical techniques especially sample storage, transportation and processing are the key factors for effective and unbiased
results. Loss of less abundant proteins, or protein modifications during repeated freeze thaw cycles, or improper storage are
known to affect the results. Use of proteomics tools for diagnosis need a special attention to these issues as such changes may lead
to error prone diagnosis.
Samples, be it plasma or tissue samples, are generally not stable at room temperature. They must always be stored frozen at -80 0C.
Even while handling them, care should be taken to keep aliquots on ice so that minimal damage is caused by repeated freeze-thaw cycles. Many authors have repeatedly stated that a majority of low abundance and/or low molecular weight proteins deteriorate
either in quality or quantity or both upon repeated freeze-thaw cycles which for some proteins(such as VEGF) is as low as 1 F/T
cycle [50].
The ideal practice to avoid sample stability issues is to make one time use aliquots of the total sample volume and prevent repeated freeze thaw cycles of the entire sample quantum [50]. Even these aliquots should be thawed on ice to prevent any damage to the sample. Vigorous vortexing of samples should also be avoided [49].
Apart from temperature, another major disruptor of stability is the proteases that maybe present on any random surface and may contaminate the sample thereby degrading it. The easiest and most convenient solution to this issue is the addition of commercially available protease inhibitor (PI) cocktails to the sample after preparing it from raw tissue or blood as early as possible.
A small yet very significant issue with samples storage is the sterility of the containers like trays, candles of cryocans, racks for -80 ˚C etc. Remnants of past samples within the containers may be a cause of contamination to the current samples. Thus such containers should either be sterilized regularly or replaced. An obvious additional precaution would be the use of sterile autoclaved tips and other accessories while handling the sample.
In conclusion, one can say that the biggest issues with sample storage and stability in proteomics are temperature and environmental proteases. Researchers can safeguard their samples against both by keeping the sample below 4 ˚C and adding PI cocktail.
Another major issue concerning sample stability and storage is sample transportation, particularly in the case of clinical samples which may require transport from the site of collection to the processing lab. In such cases samples should be immediately snap frozen in liquid nitrogen and sealed in a sterile cryocans [50]. In any case the sample must finally be kept at -80 ˚C or liquid nitrogen for long term storage.
The solvation of the protein before processing in an appropriate buffer is mandatory. The buffer provides hydrophobic additives, protease inhibitors and ions required to prevent the denaturation of the proteins present in the sample. While making a solution the protein sample concentration should be kept high (> 1mg/ml). If that isn't feasible an inert protein should be added so as to increase the concentration. The inert "buffer protein" should be easily separable from the protein of interest [51].
SOMAmers are an evolving new class of fully synthetic, single stranded DNA-based molecular recognition elements (MREs) with two key innovations. The first is a versatile set of chemical modifications that endow SOMAmers with protein-like and other functional groups [47,48]. Synthesis and generation of SOMAmers are based on in vitro screening of large libraries of randomized sequences by a modified version of the process of systematic evolution of ligands by exponential (SELEX) enrichment [49]. SOMAmer technology platform provides efficient, large-scale selection of MREs developed specifically to enable highlymultiplexed proteomic assays. SOMAmers proteomics platforms are being applied in diagnosis of Cancer, cardiovascular disease, renal disease, neurological disease, inflammatory disease, infectious diseases (www.somalogic.com).
Subcellular proteomics and study of post translational modifications are the two emerging queries that are being seeked through proteomics, although naïve but this information would be highly specific and could provide a powerful support to the upcoming personalized medicine approach. Subcellular proteomics involves techniques such as fractionation of cellular compartments and then use of conventional proteomic techniques. As eukaryotic cells are highly compartmentalized both in terms of structure and function, the exploration of sub cellular proteins would provide a way for diagnosis and discovery of therapeutic targets for organelle associated diseases such as mitochondriopathies, lysosomal storage diseases etc. Protein modification on the other hand are versatile and mostly reversible, however some of the protein modifications such as carbonylation is irreversible and could be used as possible diagnostic marker.
Recent past has witnessed a change in proteomics approaches, previously known top down approaches such as 2D Gel electrophoresis, are now considered obsolete and questioned due to several limitations. Newer approaches based on bottom up proteomics and gel free systems are emerging rapidly. Yet, in comparison to the genomic era the proteomics research needs to takes a quantum leap which can create a paradigm shift in proteomics application for diagnosis. Post genomic era would surely witness a dramatic change in the proteomics studies and many techniques that are at the doorstep of the clinical research would certainly make their way into the market. Although complexity and dynamics of proteome are biggest hurdles in the study of proteomics, yet they are most promising opportunities for the development of biomarkers and disease fingerprints in coming time. Completion of several cellular and subcellular proteomes could also be integrated into virtual cell simulation programs and this would escalate the development of point of care diagnostic techniques for pathogenic, multifactorial and metabolism associated diseases.
Arya A and Paul S are recipient of research fellowship from Council of Scientific and Industrial Research (CSIR), New Delhi. Gangwar A, is the recipient of INSPIRE fellowship from Department of Science and Technology (DST), New Delhi, India.

![]()

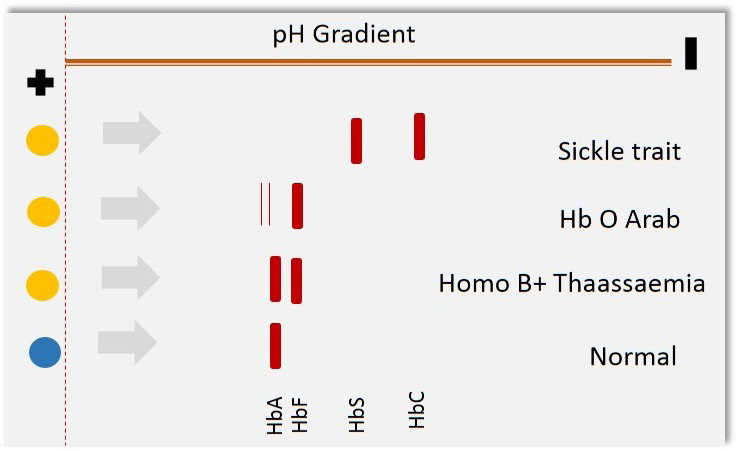 |
| Figure 1: Schematic representation of isoelectric focusing technique for disease diagnosis |
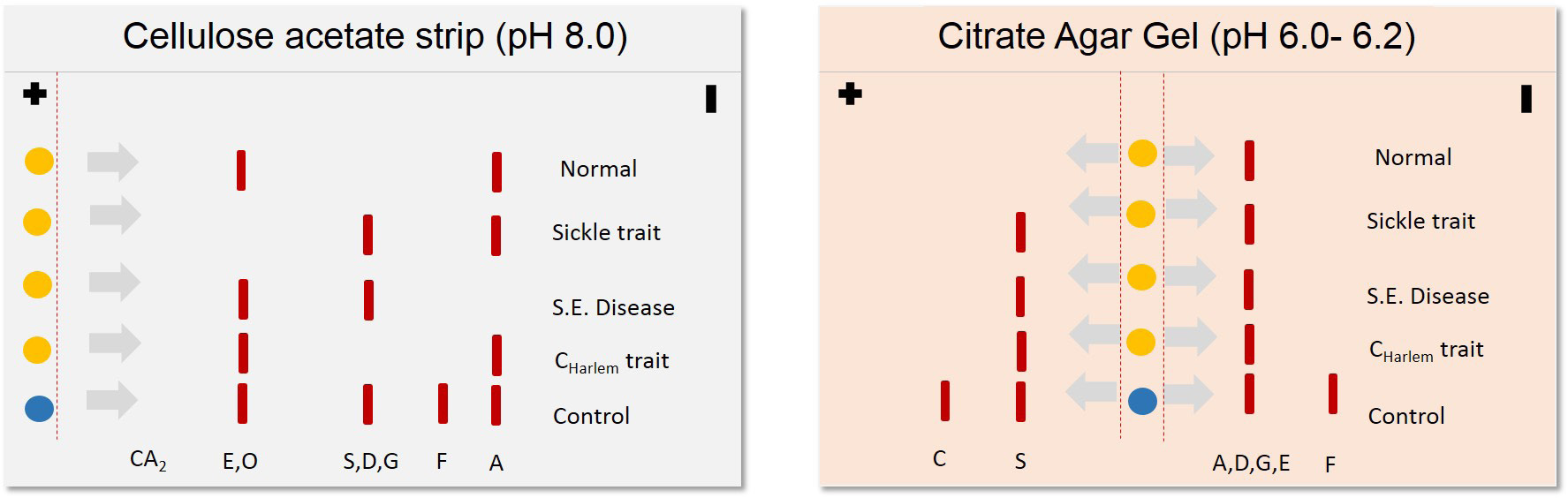 |
| Figure 2: Schematic representation of cellulose acetate and citrate agar gel electrophoresis for diagnosis of Hemoglobinopathies |
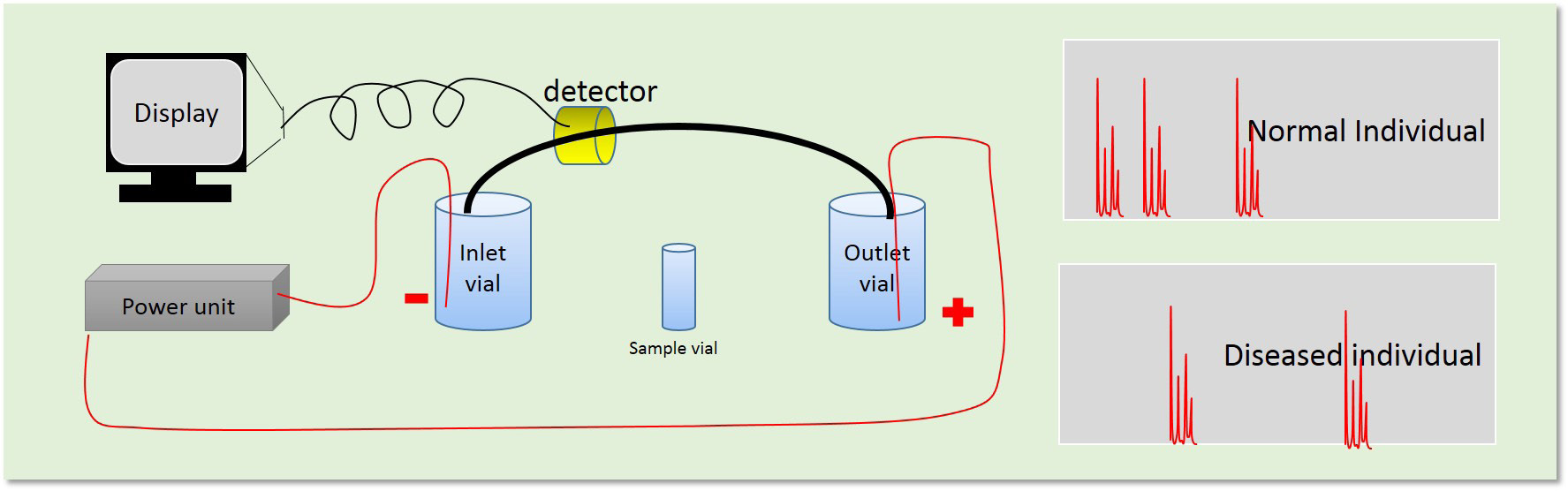 |
| Figure 3: Schematic representation of isoelectric focusing technique for disease diagnosis |
| METHOD DESCRIPTION | ADVANTAGES | DISADVANTAGES | SENSITIVITY |
| 2D GEL ELECTROPHORESIS/MASS SPECTROMETRY | |||
| ■ Separation of complex proteins via 2D gel electrophoresis based charge and size ■ Major protein identification by MS ■ Detects about 2000-2500 spots/gel |
■Ability to identify unknown proteins ■ Detects protein modification (phosphorylation and methylation) ■ Used for various biological samples, including tissue, blood and other biological fluids |
■ Proteins expressed at low abundance may be missed ■ Unsuited for diagnostic application ■ Limited reproducibility and high rate of false identification ■ Limited dynamic range ■ semi-quantitative |
■ Detection sensitivity is in the
nanogram range (50 ng/spot for Coomassie Blue; 1 ng/spot for silver stain) ■ Using fluorescent 2D-differential gel electrophoresis (2D-DIGE), sensitivity improves by 10 fold (CyDye label) |
| LIQUID CHROMATOGRAPHY/MASS SPECTROMETRY | |||
| ■ LC to separate proteins in a sample, with sequential LC for improved separation efficiency ■ MS to systematically identify the major proteins ■ Detects over 1000 proteins/run |
■ Ability to identify unknown proteins ■ Improved separation efficiency compared to 2D gel ■ Used for various biological samples, including tissue, blood and other biological fluids |
■ Proteins expressed at low abundance may be missed ■ Unsuited for diagnostic application ■ Limited reproducibility and high rate of false identification ■ Limited dynamic range ■ semi-quantitative |
■ Detection sensitivity is in the nanogram range or ~20 cells ■ 1% false positive rate |
| PROTEIN ARRAY | |||
| ■ Individual protein immobilization on a solid-support (glass or membrane) ■ Individual proteins identified by labeled antibodies ■ Detects over 1000 proteins/array |
■ High sensitivity and specificity ■ Good quantitation range ■ High throughput/density amenable for automation ■ Economical and low sample consumption ■ Lots of data from single Experiment ■ Software and hardware tools may be shared with DNA microarray |
■ Limited protein availability from complex protein production process (expression and purification) ■ Limited access to a large number of affinity antibodies for detection. |
■ Detection sensitivity is in the ng/ ml range |
| REVERSE PHASE PROTEIN ARRAY | |||
| ■ Multiple whole-cell or tissue lysate immobilization on individual spots on a solid support (similar to tissue microarray format) ■ Presence of specific proteins are detected by antibody ■ Detects < 100 proteins/array |
■ Highly sensitive detection of proteins ■ High throughput, i.e. a large number of samples on one slide ■ Minimal sample required ■ Reduced number of antibodies needed to detect protein |
■ Detection sensitivity may be compromised from loss native protein conformation when surface spotted ■ Limited sensitivity to detect low abundance proteins ■ Specificity may be compromised from non-specific antibody binding (i.e. potential for high background) ■ Limited number of available signaling protein-specific antibodies |
■ Detection sensitivity is in the picogram range ■ Increased sensitivity ■ Using laser capture microdissection, lysates can be analyzed with as few as 10 cells |
| ANTIBODY ARRAY | |||
| ■ Capture antibodies are spotted and fixed on a solid surface ■ Proteins (antigens) are captured on the array surface and detected by a second antibody specific for a different epitopes than capture antibody (sandwich format) ■ Detects < 100 proteins/array |
■ Highly specific from dual antibody detection ■ Highly sensitive ■ High throughput and amenable for automation ■ Possible to detect protein modifications (phosphorylation, methylation, etc) by modificationspecific antibodies ■ Suitable for clinical applications |
■ Protein complexity and denaturation may affect antigenantibody interaction ■ Need for high-affinity and specific antibodies for capture and detection ■ Limited dynamic range of 2 or 3 orders of magnitude |
■ Detection sensitivity is in the low pg/ml range |
| BEAD-BASED ARRAY | |||
| ■ Either capture antibody or proteins are coated on beads ■ Detection of proteins by labeled antibodies (similar to antibody array or ELISA) ■ Detects 50-100 proteins/run |
■ Highly sensitive and specific ■ High throughput and amenable for automation ■ Detects protein modifications (phosphorylation, methylation, etc) by modification specific antibodies ■ Suitable for clinical applications |
■ Protein complexity and denaturation affecting antigenantibody interaction ■ Need for high-affinity and specific antibodies for capture and detection ■ Limited dynamic range of 2 or 3 orders of magnitude |
■ Detection limit is sufficient to capture low abundance protein analytes down to the pg/ml range |
| Table 1: Key proteomic techniques in use for diagnostic applications: Advantages and disadvantages | |||
| TECHNIQUE | DISEASE DIAGNOSIS | REFERENCES |
| Cellulose acetate/citrate agar gel electrophoresis | Hemoglobinopathies | Schedlbauer LM et al., 1989 Robinson, AR. J et al.., 1957 |
| Isoelectric Focusing (IEF) | Multiple sclerosis | Andersson M. et al., 1994 |
| Capillary electrophoresis | Metabolic disease | Katja B. et al., 1997 |
| Ulcerative colitis, Crohn’s disease, | Pool, M.O et al., 1995 | |
| Scrapie(sheep) | Schmerr, M.J. et al., 1998 | |
| Turbidimetry | Lipase activity associated with pacreatitis | Shihabi, Z.K., 1970 |
| C-reactive protein associated with inflammation | Otsuji, S., 1982 | |
| Pulmonary hypertension | Brown, M.D., 2003 | |
| Table 2: Summary of proteomics techniques established in early 90’s | ||
| TECHNIQUE | DISEASE DIAGNOSIS | REFERENCES |
| Immuno-Affinity methods | HIV, Ebola, polio, Malaria autoimmunoe disorders | www.omegadioanostics.com |
| 2-D DIGE | Creutzfeldt-Jakob disease | Brechlin, P. et al., 2008 |
| MALDI-TOF MS | FOS in urine (lysosomal storage disease) | Xia, B. et al., 2013 |
| Rapid identification of various pathogenic bacterial/fungal cultures. | Patel, R. et al., 2014 | |
| LMD/MS | Renal amyloidosis/glomerulonephritis | Sethi S et al. 2013 |
| LC-MS/MS | Endocrine disorders, Vitamin D analysis | Qing H. Meng, 2013 |
| iTRAQ | Diagnosis of cGVHD-clinical trials (no. COG-ASCT0031) | Fujii, H. et al., 2008 |
| ESI-MS/MS | Inborn errors of metabolism in newborns. | Schulze, A. et al., 2003 |
| Table 3: Summary of presently used proteomics tools used in diseases diagnostics | ||
| Sr. No | Technique | Application | Advantages | Limitation |
| 1 | Isotope coded affinity tags (ICAT) | Relative quantification for protein estimation |
Sensitive and detects peptides at very low concentrations | Could not identify proteins without lysine. |
| 2 | Stable isotope labelling with amino acids in cell culture (SILAC) |
Detection of differential expression of proteins |
High degree of labelling and straightforward estimation |
Could not be done for tissue, limited to cell culture. |
| 3 | Isobaric tags for relative and absolute quantification (iTRAQ) |
Relative quantification using isobaric labelling |
High throughput, samples can be multiplexed | Increased sample complexity |
| 4 | Multidimensional protein identification technology (MudPIT) | Identification of protein- protein interaction |
Large protein complex identification | Not quantitative, High throughput is not possible |
| 5 | Protein array | Quantitation of specific proteins (usually disease biomarkers) |
Smaller sample volumes required and highly sensitive |
Limited by protein and antibody availability and high cost |
| 6 | Reverse phase protein array | Robust quantification and estimation of proteins | Less expensive and simple to perform. | Emerging, not established |
| 7 | SOMAmers Technology | Cancer, cardiovascular diseases. Pulmonary diseases |
Multiplex and high throughput | Relatively higher cost |
| Table 4: Summary of advanced proteomics techniques with prospective application in diagnosis | ||||




































