Open Access
Research Article
Max Screen >>
ISSN: 2454-4981
Copyright: © 2017 Marcon M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
It is still contentious whether amyotrophic lateral sclerosis (ALS)-Alzheimer’s disease (AD) cases comprise a combined disease or represent a coincidental association. We describe the case of a 78-year-old white male with a 12-month history of bulbar muscle weakness and mild cognitive impairment. Hallmarks of AD pathology were present and associated with diffuse congophilic angiopathy. The motor cortex did not show a loss of motoneurons. The anterior horns of the medulla oblongata and hypoglossal nuclei showed skein-like cytoplasmic inclusions and ubiquitin and TDP43 phosphate positive staining. No signs of FTLD were detected. The pathological results suggest that, in this case, ALS and AD pathology appeared to be independent.
Keywords: Alzheimer disease; Amyotrophic lateral sclerosis; Transactive response (TAR)-DNA-binding Protein 43 (TDP-43)
List of Abbreviations: AD: Alzheimer Disease; ALS: Amyotrophic Lateral Sclerosis; TAR: Transactive Response; TDP-43: Transactive response-DNA-binding Protein 4343)
The neuropathological findings of ALS consist of a severe loss of motor neurons that often contain small eosinophilic inclusions (Bunina bodies) and ubiquitin-positive filamentous inclusions (skeins) [1]. The transactivation response (TAR) DNA binding protein 43 kDa (TDP-43), a ubiquitously expressed ribonucleoprotein, is the major component of neuronal cytoplasmic inclusions (NCIs) in frontotemporal lobar degeneration with ubiquitin inclusions (FTLD-U) and ALS [2].
Initially considered relatively specific to FTLD-U and ALS, TDP-43 pathology has recently been detected in a number of other neurodegenerative diseases, including Alzheimer’s disease. Indeed, several studies have shown TDP-43 deposits in up to 30% of AD brains, particularly in the limbic system, where the amygdala seems to be the most affected portion [3-6].
These findings suggested the hypothesis that dysregulation of TDP-43 might be involved in neurodegeneration in each of these disorders and that a possible pathogenetic ground could link these different diseases. However, it is still controversial whether these cases represent a combined disease; that is, mixed AD and/or FTLD-U [7]. From this perspective, we hereby report a patient with ALS-AD who had undergone brain neuropathological examination.
A 78-year-old male was admitted with a 12-month history of progressive dysphonia and dysphagia.
At admission, neurological examination showed mild dynamic ataxia, hypotrophism of the first dorsal interossei muscle of the right hand, brisk muscular reflexes except for ankle jerk reflexes, bilateral Babinski signs, dysarthria, dysphonia and lingual fasciculations, neck muscle weakness, and fasciculations of both biceps femoris. In brief, the diagnostic workup was as follows.
Bioumoral tests were normal. Echo color Doppler of the arteries of the neck showed 50% left carotid stenosis. Brain MRI was normal except for a mild diffuse atrophy, and spinal MRI did not reveal medullary lesions. A neurophysiologic workup indicated a neurogenic pattern in the four limbs. Standard cerebrospinal fluid (CSF) was normal. CSF amyloid beta 1-42 was 135 pg/ml (n.v. >550 pg/μl); Total Tau and phosphor Tau levels were 839 pg/ml (n.v. <375 pg/ml) and 55 pg/ml (n.v. <52 pg/ml), respectively.
The patient underwent neuropsychological testing consisting of MMSE, FAB (Frontal Assessment Battery), Letter fluency test, Verbal digit span, Babcock story recall test, Raven’s Couloured Progressive Matrices, Clock drawing test and Trail Making tests A and B. The neuropsychological examination indicated multiple cognitive difficulties characterized by impaired attentional control (in particular, shifting/inhibition functions and motor planning), subtle problems in visuospatial planning and in memory recall. MMSE results were 27/30. According to NIA-AA criteria, the final report was multiple-domain MCI [8,9]. At the end of our clinical and instrumental workup, the diagnosis was probable ALS (sec. revised El Escorial Criteria) with associated multiple-domain MCI [10].
After two months, the patient died and we requested brain immunohistochemistry (ICH) to confirm ALS and to clarify the diagnosis responsible for the neuropsychological deficits and their relation to ALS.
The brain was fixed in 10% buffered formalin with subsequent processing and paraffin embedding of samples from the frontal, temporal, parietal, and occipital lobes, hippocampus, amygdala, corpus striatum, thalamus, hypothalamus, cingulate gyrus, cerebellum, mesencephalon, pons and three different levels of the cervical spinal cord. All formalin-fixed paraffin-embedded (FFPE) tissue sections were stained with Woelcke’s hematoxylin to highlight myelin; all sections were also stained with hematoxylin and eosin. TDP-43 immunostaining was performed on selected sections of the frontal cortex, temporal cortex, hippocampus and spinal cord. Paraffin sections (9μm) were deparaffinized in xylenes, rehydrated with graded alcohols and endogenous peroxidase was blocked with 3% hydrogen peroxide in PBS for 5 minutes. Heat-induced antigen retrieval was performed with citrate buffer, pH 6.0 (Sigma-Aldrich, Darmstadt, Germany) in a thermostatically controlled water bath at 95 °C for 20 minutes. After blocking with 2% normal serum (goat serum for polyclonal primary antibodies or horse serum for monoclonal primary antibodies; 30 minutes), sections were incubated overnight at 4 °C with anti-phospho TAR-DNA binding protein 43/anti- phospho-TDP-43 (rabbit polyclonal pS409/410-1, 1:2,000 or mouse monoclonal pS409/410, 1:10,000; both purchased from Cosmo Bio Co., LTD, Tokyo, Japan).
Sections were washed in PBS (3 × 10 minutes), incubated with biotinylated anti-rabbit or anti-mouse immunoglobulin G (1:200; Vector Laboratories, Burlingame, CA; 1 hour; room temperature) and treated with preformed avidin-biotin-conjugated enzyme complex (Vector Laboratories, Burlingame, CA). The staining was visualized with a NovaRED Peroxidase (HRP) Substrate Kit (Vector Laboratories, Burlingame, CA).
The slides were then counterstained with Mayer’s hematoxylin. Sections were cleared in alcohols and xylenes and coverslipped with mounting medium.
Macroscopic examination revealed only a mild bilateral reduction of cortical grey matter in the frontal, temporal and parietal lobes and no focal lesions.
In the cortical sections, neuronal density was relatively preserved but numerous senile plaques and neurofibrillary tangles were present in all cortical sections examined without a gradient of severity. ICH with anti-tau antibodies showed the presence of neurofibrillary tangles and dystrophic neuritis. ICH with anti-beta-amyloid antibodies showed the presence of ubiquitous diffuse and core amyloid plaques. Both findings were also present in the central grey nuclei, claustrum, thalamus and mesencephalon (Figure 1a). Almost all parenchymal and meningeal vessels were immune positive for amyloid deposition (congophilic angiopathy) (Figure 1b). The neuropathological feature described above appears typical for AD. In the brain sections, we did not find TDP-43 pathology.
The motor cortex did not show a loss of motor neurons, and the pyramidal tracts showed normal staining for myelin with Woelcke’s hematoxylin. In the cervical spinal cord (Figure 1c) and in the medulla oblongata, we found typical pathological features of ALS, characterized by degeneration and loss of motor neurons in the anterior horn of the spinal cord and in the lower cranial motor nuclei of the brainstem and, in particular, at the level of the hypoglossal nuclei (Figure 1d), as seen with routine stains such as hematoxylin and eosin (H&E). Furthermore, the same regions displayed skein-like cytoplasmic inclusions and ubiquitin and TDP43 phosphate positive staining. No signs of frontotemporal degeneration were detected.
The conclusions of the study are Alzheimer’s degenerative disease was present in an advanced state. We observed the presence of cytoplasmic inclusions and ubiquitin and TDP43 positive staining, which was compatible with ALS.
The regional distribution of AD hallmarks described an advanced stage of the disease (Braak stage IV). AD hallmarks in ALS without dementia cases have been reported as relatively rare, although one study found analogous levels of AD pathology in a group of 20 age-matched non-demented ALS cases compared with 17 non-demented controls [8-12]. Amador-Ortiz et al. demonstrated that as many as 20% of AD cases have TDP-43 pathology in the amygdala and hippocampus, suggesting that these cases might represent a combined disease; that is, mixed AD/FTLDU [4]. Josephs et al. found abnormal TDP-43 immunoreactivity associated with a modified clinicopathologic and radiologic phenotype in 34% of an AD sample [5]. In our case, FTLD pathology was absent in the brain sections and the TDP-43 pathology was confined to the anterior horn of the cervical cord and to motor brainstem nuclei. In a more recent study, it was demonstrated that frontal TDP-43 is not always the case in AD, suggesting that abnormal TDP-43 deposition and AD pathology (formation of senile plaques and neurofibrillary tangles) might occur independently [7]. In our case, the hypothesis of the coexistence of the AD pathology was generated by the results of the CSF biomarkers, whereas the neuropsychological tests suggested an executive dysfunction, characteristic of FTLD. Another way to confirm this diagnosis in vivo could be through the use of amyloid PET, which at that time was unavailable in our department.
Our CSF results were highly suggestive of AD according to the recent finding that a CSF AD-like profile in FTD is associated with brain abnormalities at 99mTc-ECD SPECT, which is typically found in classical AD, confirming the usefulness of CSF testing [13].
Autopsy procedures were carried out only on a minority of patients who died of ALS, which raises the possibility that independent association of ALS/AD pathology, as demonstrated in our patient, could be underestimated. More research is needed to understand how TDP-43 contributes to degeneration in the context of ALS, AD and other known neuropathologies.
This case report indicates that the distribution of ALS pathological hallmarks was in agreement with the clinical presentation, whereas pathological results suggested that ALS and AD pathology appeared to be independent.

![]()

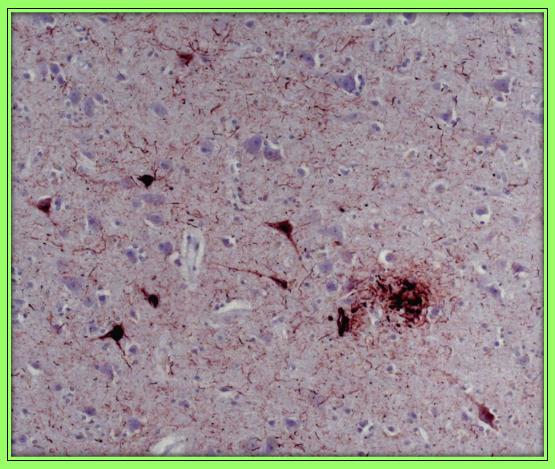 |
| Figure 1a: Neurofibrillary tangles and swollen dystrophic neurites that form the outer rim of the senile plaques. ICH stained with monoclonal Thermo scientific, clone AT8 for tau, directed against abnormally phosphorylated tau |
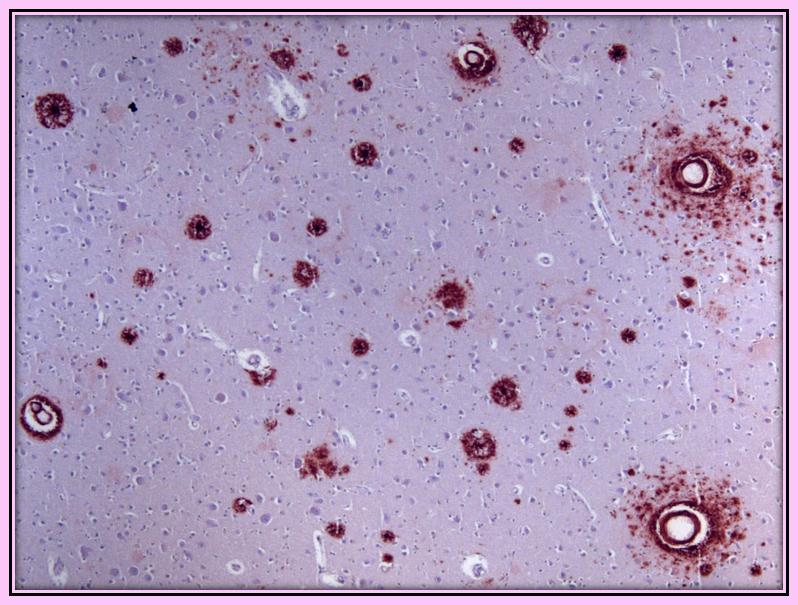 |
| Figure 1b: Cortex’s section with numerous senile plaques and amyloid in vessels walls stained with monoclonal AB Dako clone 6F/3D for beta-amyloid |
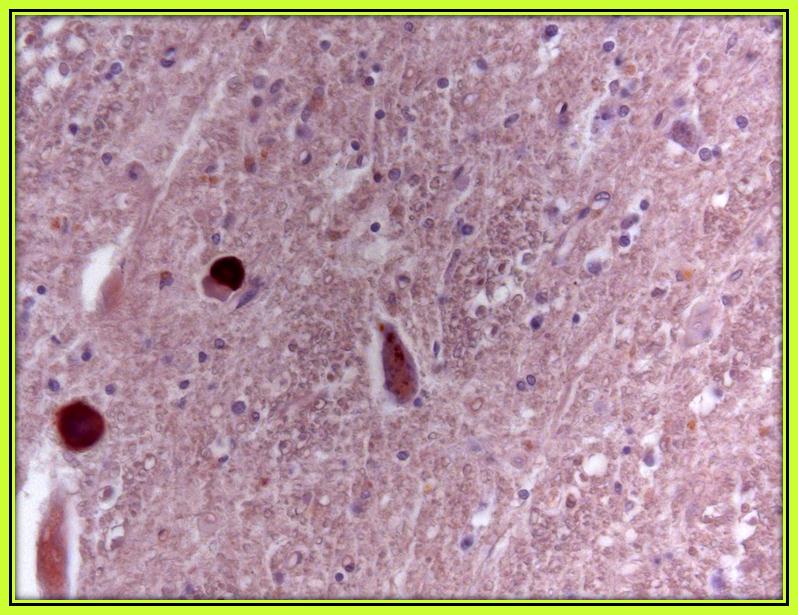 |
| Figure 1c: Nuclear staining of TDP-43 was detected consistently in motor neuron of cervical spinal cord with TARDBP monoclonal Ab-nova, clone 2E2-D3 for TDP43 |
 |
| Figure 1d: TDP 43 expression was also detected in medulla tissues with polyclonal CosmoBio for TDP43 Phospor |




































