Open Access
Case Report
Max Screen >>
ISSN: 2455-7641
Copyright: © 2016 Dongre T. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Down syndrome or constitutional trisomy 21 was linked to leukemia for the first time in a case report published in 1930 since then, down syndrome has been recognized as one of the most important leukemia – predisposing syndromes and patients with down syndrome and leukemia have unique clinical features and significant differences in treatment response toxicity profiles compared to patients without down syndrome.
Keywords: AML-M7; AML NOS; Down syndrome
Patients with down syndrome have a higher risk (10 to 20 fold) of developing leukemia [1,2]. There is approximately 150-fold increase in acute myeloid leukemia in Down Syndrome (DS) children less than 5 years of age; 70% of the cases of AML in Down Syndrome children, less than 4 years of age, are acute megakaryocytic leukemia, in contrast to the 3-6% incidence of this form of leukemia in non-DS chidren [3]. In the WHO classification of tumors of hematopoietic and lymphoid tissues 2008, myeloid proliferations related to down syndrome has been incorporated as a distinct entity. We report a case that morphologically appeared to be megakaryocytic leukemia which on flow cytometry confirmed as AML –NOS. We report a case of rare association of AML-NOS, with down syndrome with sickle cell trait disease.
A 2.5 kg female child was born to a 23 years old mother of a non-consanguineous marriage by normal spontaneous vaginal delivery. Ultrasonography was done at third month, was suggestive of increased nuchal translucency, but mother was not willing to abort. There was no history of drug intake or exposure to radiation during the antenatal period. At birth the baby has mongoloid features with upward slant of eyes, low set ears, hypertelorism, macrognathia, hypotonia, low hair-line, pre-auricular tag. On day 3, the baby was admitted to NICU for jaundice, phototherapy was not given as the bilirubin was not much raised. The complete hemogram at this time was within normal limits indicating that there was no suspicion of Transient Myeloproliferative Disorder. Breast feed was started within half hour of delivery, but was started on top feed because of decreased secretion of mother. There was no evidence of sepsis. She was discharged with close follow-up. The baby was weaned and was taking most of the regular food made at home. At the age of one and half years she presented with the complaint of diarrhea. Clinically the patient had fever since 10-15days, loose stools since 2 days, general examination of the patients revealed cardiovascular, respiratory system within normal limits. There was no evidence of any organomegaly or lymphadenopathy. Developmental milestones were little delayed (not able to walk at 18 months of age). The routine investigations revealed hemoglobin 3.6gm/dl, total leucocyte count 20000/cmm, with neutrophils 20%, lymphocytes 32% and blasts cells 48%, the blasts were large with increased nucleo-cytoplasmic ratio and agranular cytoplasm. Platelet count was 0.85 lac/cu mm. Peripheral smear report was given as two differential diagnosis as Transient Myeloproliferative Disorder and megakaryoblastic leukemia, as the child was of down syndrome and the blasts cells shows pseudopod formation (Figure 1). However EDTA blood sample was sent for flow cytometry for confirmation. However, the patient was not willing to take chemotherapy and was discharged after giving symptomatic treatment. The patient got admitted thrice after she was diagnosed. First time, two months after been diagnosed , for fever, at this time she developed moderate degree of hepatosplenomegaly, the complete hemogram was total leucocyte count 28900/cmm, differential count was blasts 64%, neutrophils 14%, lymphocytes 22%, eosinophils 00%, monocytes 00%, platelet count 1.19 lacs/cmm, hemoglobin 10.1gm/dl. Second time the patient again presented two and half months after, for same complaint, fever, with hepatosplenomegaly, the total leucocyte count was 68,000/cmm, blasts cells 70%, neutrophils 17%, lymphocytes 12%, eosinophils 00%, monocytes 01%, platelet count 1.19lacs/cmm. The blasts count of the patient is steadily increasing and the patient is frequently been admitted for fever. Third time the patient again was admitted for fever, with gross hepatomegaly and moderate splenomegaly, at this time the complete hemogram was hemoglobin 5.8gm%, total count 51000/cmm, blasts 68%, neutrophils 12%, lymphocytes 18%, eosinophils 00%, monocytes 02%, at this time the peripheral smear showed occasional sickle-like cells (Figure 2). Hemoglobin electrophoresis was done, which confirmed it as AS pattern of sickle cell disease. The patient remained alive for almost six months after been diagnosed, and was managed symptomatically. She was never started on chemotherapy, as the parents were not willing for it. On third admission, the patient expired.
Histological examination of the bone marrow shows replacement by same type of blasts as seen in peripheral smear, so it made flow cytometry mandatory for confirmation.
AML –NOS encompasses those cases that do not fulfill the criteria for any of the other AML categories this category accounts for only 25% to 30% of all cases and it will continue to diminish as more genetic subgroups are recognized.
Flow cytometry report was positive for CD 34, CD 117, CD7, CD56, moderately positive for HLA-DR, CD 13, CD33 and CD36. Blasts were negative for CD41, CD61, CD14, CD64, cytoplasmic CD3 and other B and T-cell markers (Figure 3).
Based on the flow cytometry report, it was confirmed as acute myeloid leukemia (AML-NOS) with aberrant CD7 expression.
A unique syndrome occurs only in newborn infants with Down Syndrome referred to as transient myeloproliferative disorder (TMD), transient abnormal myelopoiesis (TAM), or transient leukemia (TL). In addition, children with Down Syndrome are also at increased risk for acute megakaryoblastic leukemia (AMKL) and acute lymphoblastic leukemia (ALL) in the first 4 years of life [2]. When treated with chemotherapy, children with DS also experience more side effects (such as infections and mucositis) from treatment than children without DS [2-6]. Remarkably, children with DS AML have a better prognosis than children with non-DS AML. In addition, their clinical characteristics differ from those of non-DSAML children: younger age, lower white blood cell count (WBC), and high incidence of French-American-British (FAB) M7.
Trisomy 21 is considered the first genetic event in DS-AMKL leukemogenesis, the second hit is a mutation of the X-linked gene GATA1, encoding a blood – specific transcription factor essential for development of the erythroid and megakaryocytic lineages [5].
The first insight into the mechanism of DS-AMKL was the discovery of the acquired mutations in the GATA1 gene. These mutations were restricted to the leukemic clones and were not found in normal remission samples. The mutation is not detectable in non-DS leukemia or other subtypes of DS leukemia, emphasizing the specific cooperation of GATA1 mutation with trisomy 21 in megakaryocytic leukemia [6]. Although AMKL and TMD is the most common hematological abnormality found in patients of Down syndrome, other malignancies like AML –NOS can also be evident in these patients, indicating the importance of flow cytometry in every case of down syndrome with hematological abnormality. Significance of CD7 positive cases is that the overall survival and disease free survival rate was lower than those of CD7 negative patients. The remarkable prognosis in these patients as compared to Non Down AML reflect differences in cellular drug resistance, pharmacokinetics, or regrowth potential of residual disease. DS AML cells were significantly more sensitive to cytarabine (median, 12-fold), the anthracyclines (2-7– fold), mitoxantrone (9-fold), amsacrine (16-fold), etoposide (20-fold), 6-thioguanine (3-fold), busulfan (5-fold), vincristine (23-fold), and prednisolone (more than1.1-fold), than non-DS AML cells disease [7]. Our patient has a rare association of down syndrome with AML-NOS, with sickle cell trait.
This patient has a classical morphological picture of blasts with pseudopod formation in peripheral smear as well as in bone marrow creating a strong suspicion of AMKL, but flow cytometry confirmed it as AML-NOS. There is a wide spectrum of hematological malignancies in children with DS. The association of hemolytic disease with leukemias is rare in down syndrome. A careful scrutiny of peripheral blood film and a close follow-up is warranted to diagnose association with hemolytic disease. Although TMD and AML-M7 are the most common hematological abnormality found in patients with down syndrome, other type like AML –NOS should also be kept in mind and flow-cytometry should be done in every patient of DS.

![]()

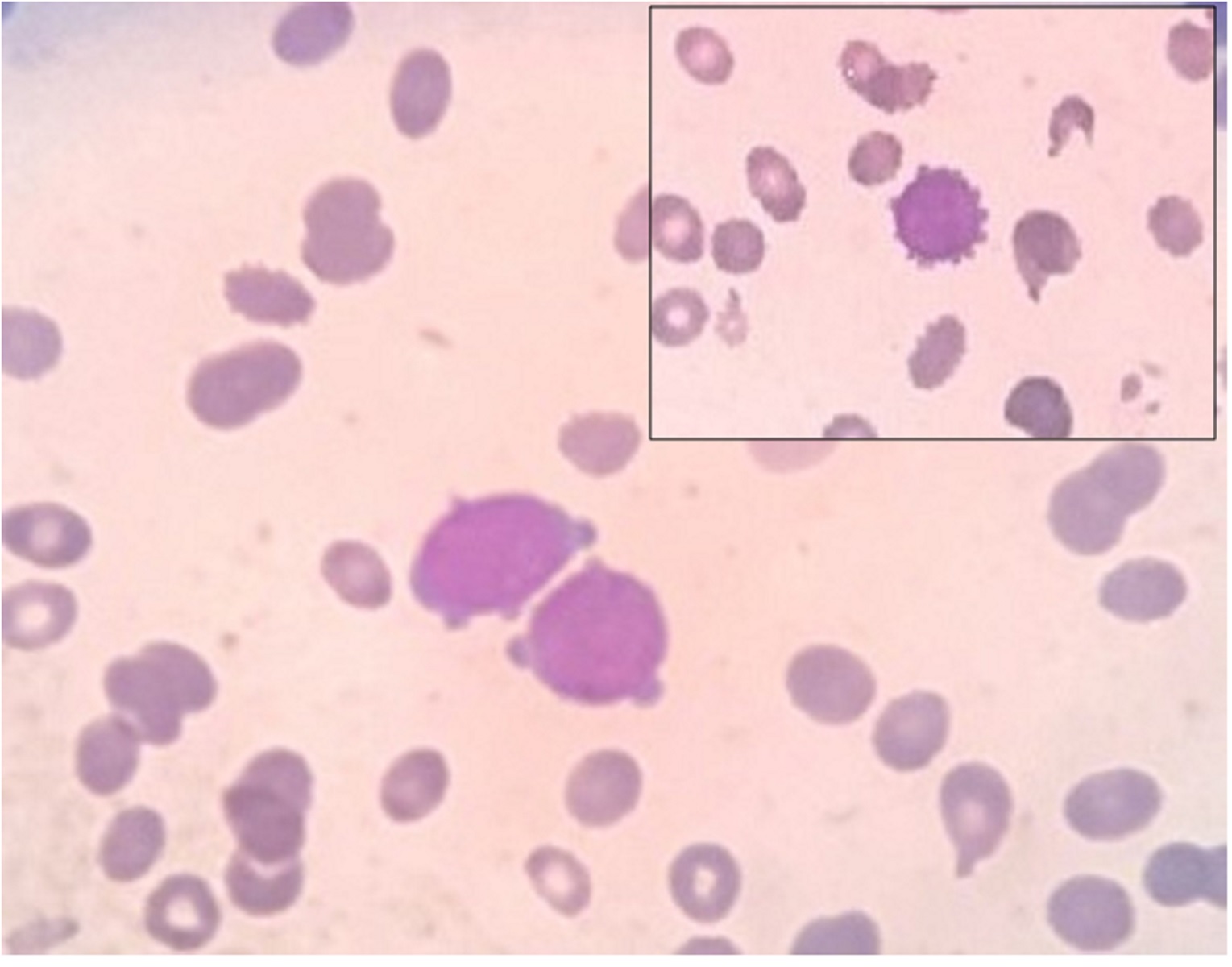 |
Figure 1: Peripheral smear showing blasts cells with pseudopod formation (400x) |
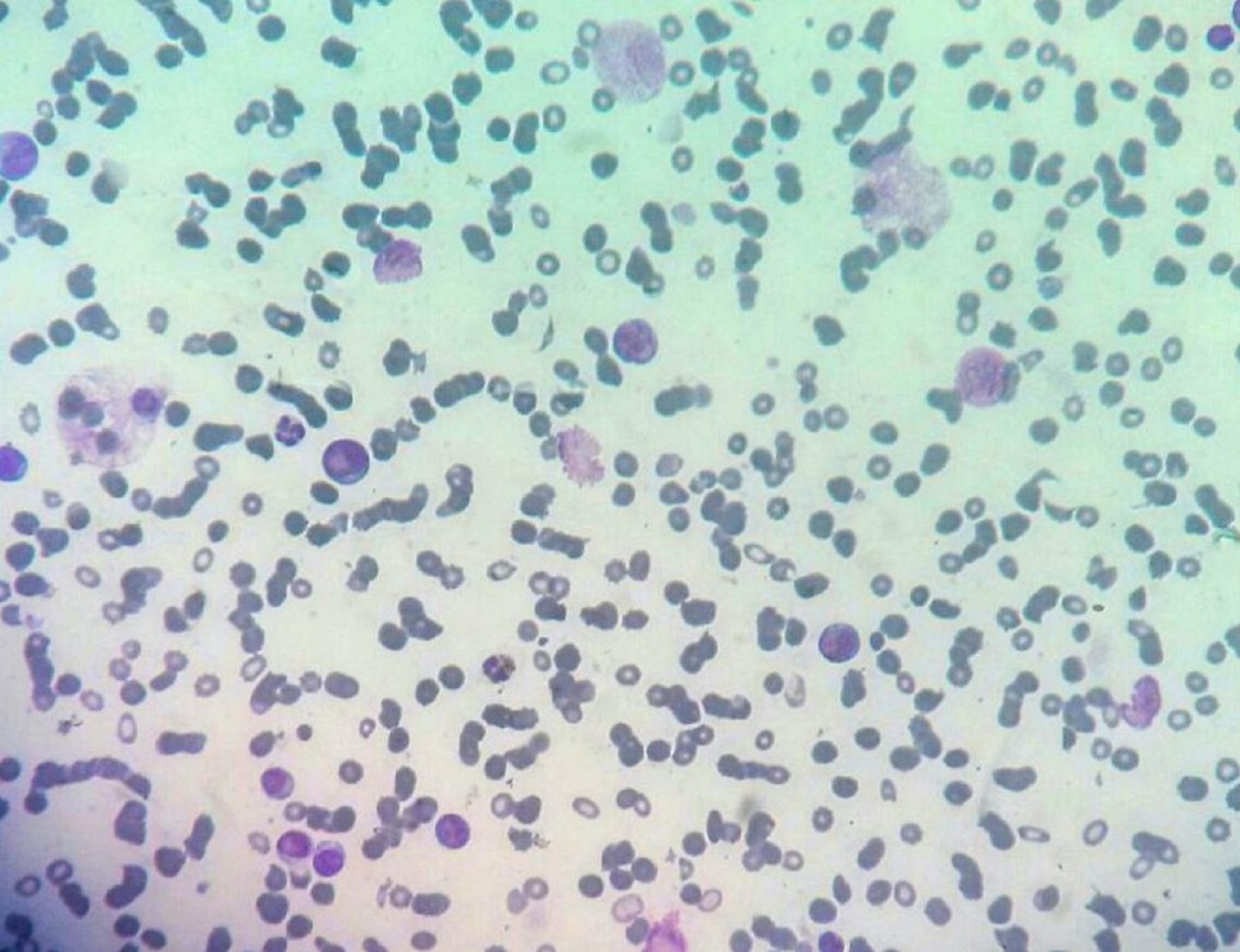 |
Figure 2: Bone marrow aspiration showing blasts cells with pseudopod formation (400x) |
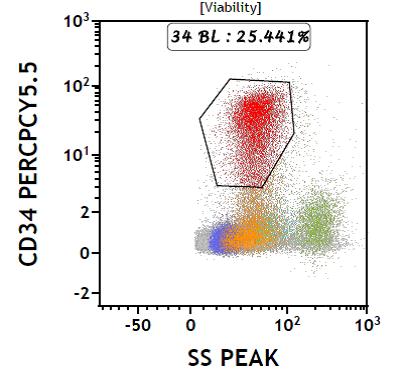 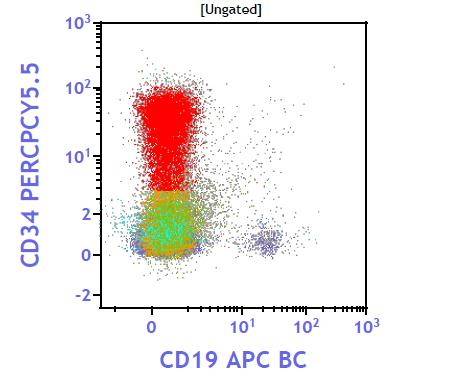 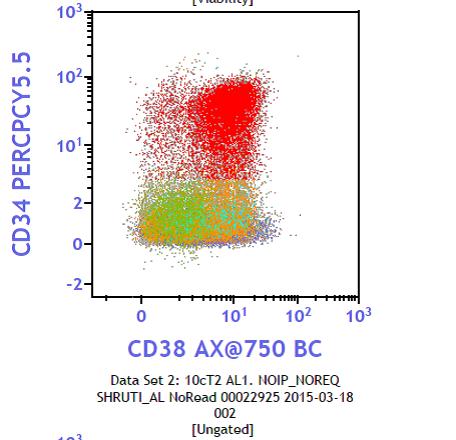 |
Figure 3: Flow chart negative for CD41 and CD61 |




































