Open Access
Case Report
Max Screen >>
ISSN: 2455-7641
Copyright: © 2016 Santiago Andrés Guzmán – García. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
A 22 year-old male with an unspecified family history of hematologic diseases was admitted complaining of chest pain. A diagnosis of thrombosis of supra-aortic vessels and hereditary spherocytosis was established and, because there is not a causal relationship between these two disorders, an approach intended to rule out primary or secondary thrombophilia is herein mentioned.
Keywords: Hereditary spherocytosis; Primary thrombophilia; Thrombosis of atypical site; Antiphospholipid antibody syndrome
List of Abbreviations: HS: Hereditary Spherocytosis; AFA: Antiphospholipid antibodies
Hereditary spherocytosis is part of a group of disorders of the erythrocyte membrane such as hereditary stomatocytosis, eliptocytosis and ovalocytosis which mainly manifest as a regenerative hemolytic anemia. Its etiology comes from genetic alterations of membrane and structural proteins which disturb the erythrocyte cohesion and stability. HS also known as Minkowski Chauffard's Disease, has a prevalence of 1 in 2000-3000 patients in north America and north Europe however a great number of cases remain asymptomatic [1]. This heterogeneous disease is characterized by spherical-shaped erythrocytes seen in the peripheral blood smear [2], which have a reduced physiological strain capacity due to anomalies of well identified membrane proteins such as ankirin, alpha y beta espectrin, band 3 and protein 4.2, this leads to a wide spectrum of clinical presentations varying in severity and age of onset [3]. In general, alpha spectrin anomalies have an autosomic recessive inheritance while beta spectrin has an autosomic dominant pattern which influences the severity of disease. The more common altered protein is ankirin representing 40-65% of all cases [4]. HS can manifest at any age but typically in adolescents and young adults; severe cases manifest at birth. Clinical manifestations include but are not limited to hemolytic anemia, splenomegaly, jaundice, bilirubin gallstones, increased osmotic fragility 50 and family history of disease [5]. Diagnosis is based in the last two in addition to mean corpuscular hemoglobin concentration (MCHC) >36 g/dl and flow cytometry with dye eosin 5 -maleimidin (EMA) which show the loss of normal erythrocyte Surface, Cryohemolisis or osmotic fragility test [2]. Treatment is determined by the severity of disease and risk-benefit of splenectomy [6]. Below we describe a clinical case of a 22 year-old male patient with HS and a venous thrombosis in an atypical site, in which, after a diagnostic approach we suspect of a triple hematologic condition: hereditary spherocytosis, primary thrombophilia and antiphospholipid antibody syndrome.
There are no reported cases of spontaneous deep venous thrombosis in patients diagnosed with HS.
The S and C proteins are vitamin K-dependent glycopoeins that serve as part of the regulston system of the coagulation cascade. Its deficiency leads to increased generation of thrombin secondary to the absence of inactivation of activated V and VIII factors. Acquired deficiency of this proteins, can be functionally divided in 2: Increased consumption or decrease in their synthesis; the first is due to pharmacological inhibition by vitamin K antagonists or due to a severe depletion of vitamin K, as well as chronic liver disease which can be demonstrated in patients with end-stage cirrhosis. None of these conditions occurred in this patient; however acquired etiologies are more frequent than congenital, with an estimated prevalence of 0.3% and 0.1% for protein C and S deficiency respectively [7].
A 22 year-old male patient presents to the clinic with a 5-day history of progressive thoracic oppressive pain, radiated to the left jaw. His personal medical record is irrelevant except by history of neonatal jaundice. He has family history of hematologic conditions; his great grandmother and grandmother were splenectomized of unknown cause, and his mother has recurrent anemia. Physical examination reveals generalized jaundice and massive splenomegaly; the inferior border of the Spleen is palpated three cm above the left anterior superior iliac spine, surpassing the abdomen midline. A Doppler ultrasound was performed revealing an intraluminal filling defect of the internal jugular vein, compatible with thrombus. Extended evaluation with a CT angiography showed thrombosis of the left internal jugular, innominate and subclavian veins, splenomegaly of 24 cm on its sagittal axis and no images compatible with pulmonary thromboembolism (Figure 1, 2a, 2b, 3 and 4). Laboratory tests show normocytic normochromic anemia with a wide red cell distribution width (RDW), prolonged activated partial thromboplastin time (aPTT), indirect hyperbilirubinemia and high levels of lactate dehydrogenase (Table 1). Peripheral blood smear showed few spherocytes, ghost cells, macrocytosis and moderate microcytosis and ovalocytosis (Figure 4 and 5). Anticoagulation with enoxaparin (1mg/kg) was started. Because of this findings and the suspicion of HS, diagnostic approach was complemented with an acute viral hepatitis panel and ELISA for HIV with non-reactive results, positive osmotic fragility test compatible with the diagnosis of HS and a mix study with non-correction of aPTT (Table 1).
An isolated prolonged aPTT result with a normal prothrombin time result implies either the presence of heparin, an inhibitor (either specific factor or lupus anticoagulant), or a factor deficiency of the intrinsic system. A mixing study may be necessary to determine the etiology. To perform it, the patient specimen is mixed with an equal volume of normal platelet free plasma, and another volume mixed with an equal volume of veronal buffered saline. APTTs are performed on each of these mixtures. An aliquot of the normal plasma/patient plasma mixture is also incubated at 37 oC for one to two hours and then an aPTT performed to further clarify the cause of the prolongation As a control, normal platelet free plasma and patient plasma are also incubated separately and then mixed after this incubation as a control for possible factor degradation during incubation.
If the result obtained with the immediate normal plasma mixture corrects to within the reference range and the saline mixture result increases dramatically, a factor deficiency or specific factor inhibitor is suspected. With factor deficiency, heparin or a specific factor inhibitor, the 1:1 saline mix shows significant prolongation of the aPTT result. If the result obtained with the immediate normal plasma mixture shows only partial or no correction and the result obtained with the saline mixture shows near correction to the reference range or only mild prolongation, then a nonspecific inhibitor such as lupus anticoagulant (LA) is suspected. After incubation at 37 oC, an aPTT result equivalent to that of the original mixture is indicative of a factor deficiency or heparin. If the result of the APTT on the incubated mixture demonstrates further prolongation (with the control remaining equivalent to the original result), a time dependent specific factor inhibitor is indicated. A specific FVIII inhibitor may show initial correction of the original mixture, but will demonstrate prolongation upon incubation. Factor deficiencies should be further identified by performing specific factor assays. Plasma suspected of containing a lupus anticoagulant (LA) should be further tested with other LA specific tests [i.e., Dilute Russell Viper Venom Time (DRVVT), Platelet Neutralization Procedure (PNP), or the Hexagonal Phospholipid Neutralization Procedure] to confirm the presence of a lupus anticoagulant.
Russell’s viper venom activates the coagulation system by directly activating FX. In the DRVVT, the term dilute refers to the reduced concentration of phospholipid used in the reagent, to increase sensitivity for Lupus Anticoagulants (LA). Lupus anticoagulants typically cause prolongation of the dilute Russell’s viper venom time. The DRVVT may be advantageous over the aPTT as a screen for LA in some instances, as this reaction does not involve FVIII. FVIII can elevate as an acute phase reactant and shorten the aPTT, diminishing the degree of prolongation seen with some lupus anticoagulants. DRVVT Confirm is a DRVVT assay which utilizes an increased phospholipid concentration. The DRVVT assay is performed in the presence of dilute phospholipid and concentration increased to show phospholipid dependence. A ratio of screen/confirm is reported [4].
Being a young patient with no known risk factors for venous thrombosis and with a mix study with non-correction of aPTT, a workout for evaluating the presence of a primary thrombophilia was started. Antiphospholipid antibody syndrome panel showed the presence of lupic anticoagulant and IgG anticardiolipin antibodies; low activity of proteins C and S and negative G20210A prothrombin gene and Leiden V factor mutation.
During his hospitalization, he had adequate response to anticoagulation treatment with good clinical improvement, so bridging anticoagulation was performed with warfarin and was discharged. He received anticoagulant treatment during 3 months with resolution of thrombosis and was evaluated ambulatory 6 weeks later with coagulation tests to confirm the persistence of the anomalies previously documented.
a. Spherocytes in peripheral blood smear
b. Jaundice and anemia
c. Laboratory test: osmotic fragility test, cryohemolysis or EMA test
e. All of the above
Correct answer: E
HS clinical presentation is heterogeneous: Clinically (asymptomatic, mild, moderate and severe disease) biochemically and genetically. Infections (Parvovirus B19, Citomegalovirus o Gastroenteritis) and other clinical states such as pregnancy can exacerbate the condition of asymptomatic patients or those with a mild disease form [1].
During the diagnostic approach, is important to inquire about family history, look and ask for the presence of typical manifestations such as jaundice, anemia and splenomegaly. Look pointedly for characteristic laboratory test results like normocytic normochromic anemia, reduced MCV, high MCHC in addition, spherocytes and reticulocytes in the PBS. With the previous data we can integrate the diagnosis, without performing other tests [2]. Nevertheless, when an accurate diagnosis can't be made we can perform confirmatory test such as glycerol lysis test, acidified glycerol lysis time and pink test; which determine the extension or percentage of lytic erythrocytes suspended in glycerol. Osmotic fragility test which determines the concentration of sodium chloride at which erythrocytes break at 50% and EMA flow cytometry test which binds to membrane proteins and determines the amount of fluorescence of erythrocytes with flow citometry. If the diagnosis is equivocal, a screening test with high predictive value for HS is helpful. The recommended screening tests are the cryohemolysis test and EMA binding (Table 2).
Our patient presented with jaundice, splenomegaly, family history of hematologic conditions and laboratory tests with microcytic hypochromic anemia with spherocytes in the PBS, which oriented the diagnostic approach. After this, an osmotic fragility test was performed, resulting in initial hemolysis with 0.85% hypotonic solution and complete hemolysis with 0.55% solution, confirming the diagnosis of HS. Other test can be used to confirm the diagnosis, like glycerol acidification lysis test or the pink test, though their sensitivity varies being 68% in the glycerol acidification lysis test, 61% in the osmotic fragility test, and 91% in the Pink test. As an alternative, we can ask for an EMA flow cytometry test being positive in >21% of cases with a sensitivity of 92.7% and specificity of 99.1% [3].
With the previous data, our patient can be catalogued with a moderate disease because he had an hemoglobin >8.0 g/dL, 16% reticulocytes, high osmotic fragility and no need of transfusions.
a. Medical management with annual monitoring and folate supplementation.
b. Surgery
c. Steroids
d. A y B
Right answer: D (Medical management and Surgery)
Clinical management of hereditary spherocytosis is individualized according to severity; considering folate supplementation (5mg day) in mild and moderate cases and surgical management with splenectomy in severe cases with the purpose of reduce hemolysis, its complications and transfusion requirements [4]. When splenectomy is considered, vaccination against encapsulated microorganisms is mandatory. Cholecystectomy can be performed in cases of coexisting gallstone disease in patients of above 10 years-old [5].
British guidelines recommend that splenectomy technique depends on the surgeon training, although a laparoscopic approach is associated with less post-surgical pain, intrahospital stay and better aesthetic results. Partial splenectomy confers minimum postsurgical sepsis risk but is associated with recurrence of hematological complications. Several studies evidence that total splenectomy does not increase the postsurgical sepsis mortality [2].
A patient with HS should be monitored annually with CBC, liver and gallbladder ultrasound or iron profile test depending on the severity of disease [3]. For the mentioned above, our patient should be monitored annually with laboratory tests according to severity and folate supplementation. In severe cases, where symptomatic thrombocytopenia or splenic infarction, splenectomy can be considered.
a. Recurrent venous or arterial thrombosis associated to an autoimmune disease
b. Presence of moderate or high levels of antiphospholipid antibodies in two or more occasions separated by at least 12 weeks and two clinical criteria
c. At least one clinical criteria and at least one laboratory criteria with moderate or high levels in more than two occasions, separated by at least 6 weeks
d. At least one clinical criteria and at least one laboratory criteria with moderate or high levels in one occasion
Correct answer: B
Antiphospholipid syndrome is characterized by the presence of venous or arterial thrombosis, pregnancy specific diseases and the presence of antiphospholipid antibodies [6].
Antiphospholipid antibodies are directed against phospholipids-binding plasma proteins, and its presence can be demonstrated by ELISA. IgG and IgM anticardiolipin antibodies present at moderate or high blood levels in two or more occasions, separated by at least 12 weeks and lupus anticoagulant detected in blood in two or more occasions separated by at least 12 weeks, according to the guidelines of the International Society on Thrombosis and Hemostasis [7]. The definitive diagnosis of antiphospholipid syndrome requires the presence of at least one of the clinical criteria and at least one of the laboratory criteria. There are no limits intervals between clinical events and the positive findings in the laboratory. Antiphospholipid antibodies that are not included in the diagnostic criteria are: anticardiolipin IgA antibodies, anti B2-glycoprotein I antibodies and antiphospholipid antibodies directed against other phospholipids.
In the case of our patient, he presented with subclavian, left brachiocephalic and ipsilateral internal yugular thrombosis, in addition, prolonged aPTT was found in the laboratory tests. Lupus anticoagulant and anticardiolipin antibodies in a titer IgG> 560, confirmed the diagnosis.
a. Anticoagulation with anti-vit K for 6 months (INR 2-3)
b. Low doses of ASA with hydroxychloroquine until low levels of antiphospholipid antibodies are documented
c. Anticoagulation with anti-vit K (INR 2-3) for 5 years
d. Anticoagulation with anti-vit K (INR 2-3) for undefined time
Correct answer: D
Before starting the treatment, its necessary to consider that carriers of antiphospholipid antibodies (AFA) do not have an homogeneous risk of thrombosis; several parameters must be taken into account to accurately stratify the risk of developing a vascular event.
1. AFA profile: Each AFA profile gives a characteristic thrombotic risk. Among the AFA, lupus anticoagulants (LA) have the strongest predictive value for clinical vascular events, increasing the risk of thrombosis by four [8,9].Thrombotic risk increases with the number of positive tests for LA. According to the classification criteria, patients with antiphospholipid antibody syndrome can be stratified into four categories depending on the number of positive AFA tests: Class I includes patients with more than one positive result in any combination, while patients with a single positive test should be classified in category II.
2. Associated autoimmune disorders: Patients with underlying systemic autoimmune disease have an increased vascular mobility. In particular, positive AFA, have been identified as one of the main determinants of thrombosis among patients with systemic lupus erythematosus [10,11].
3. Cardiovascular risk factors: Hypertension has become an independent predictor for presenting a first thrombotic episode in AFA carriers. That’s why a careful evaluation of cardiovascular status is recommended: Age, diabetes, hypertension, dyslipidemia, obesity, smoking, sedentary lifestyle, hyperhomocysteinemia, protein C and protein S, ATIII deficiency, Factor V Leiden mutation, prothrombin time, immobilization, prolonged surgery or procedures and the use of estrogen replacement therapy [12].
4. Site of thrombosis: Patients, whose first thrombosis event is arterial, are considered at higher risk of recurrence, in the same arterial distribution [13]. Treatment of AFAS remains controversial; any therapeutic decision could result in excessive or insufficient anticoagulation. The indication of indefinite anticoagulation is consensual, but its intensity and possible interruption are discussed. The new anticoagulant agents (Rivaroxaban and dabigatran), are still being studied in patients with AFAS and its use is not yet recommended. Pharmacological agents used in the treatment of thrombotic AFAS counteract the effects of AFAS preventing coagulation, including antiplatelet agents such as aspirin at low doses. Anticoagulant drugs include vitamin K antagonists (VKA), heparin and its derivatives. Treatment with AVK presents several difficulties. These agents have a slow onset of action, narrow therapeutic range and require frequent monitoring with INR. They interact with a great number of foods and drugs including immunosuppressive agents. AFA detection is affected with the use of VKA, limiting AFAS diagnosis [13]. The efficacy of heparin in the AFAS treatment is not only attributable to its anticoagulant action. Heparin interacts directly with β2GPI, diminishing its prothrombotic activity [14]. Bleeding risk: The risk of bleeding in patients with INR of 2.0-3.0 is 0.8-1.6%. The mortality due to thrombosis is higher than the death rate due to bleeding [15]. Patients with positive AFA without SLE are considered at high risk of thrombosis, so they should use low-dose aspirin. Patients with SLE and positive AFA should use hydroxychloroquine and low-dose aspirin. Patients without SLE who have experienced a primary non-cardioembolic stroke with a low risk AAF profile and modifiable risk factors should be considered for antiplatelet therapy. Patients with definite AFAS, who have presented thrombosis, should maintain antithrombotic medication indefinitely. Patients with primary venous thrombosis with a low risk profile, plus a known risk factor may have a limited duration of anticoagulation treatment for 3-6 months. Events of venous thrombosis. The initial therapy consists of unfractionated heparin or low molecular weight along with anticoagulant agents with at least 5 days (bridge therapy), seeking an INR 2.0-3.0, for an indefinite period, The persistence of anticardiolipin antibodies after 6 months of a venous thrombosis; predict thrombotic recurrence and death [13] Arterial thrombotic events: The patients with history of arterial thrombosis and AFA should use warfarin for an indefinite time (INR 2-3 or INR >3) with or without antiplatelet agents. Although prospective studies have not found any difference between INR 2-3 or INR >3, making difficult to take any definitive conclusion. We suggest long term anticoagulant therapy with warfarin intensive therapy. Additional therapeutic options: Hydroxychloroquine, which has been proved to have immunomodulatory effects; preventing the activation of TLR3, TLR7 and TLR9. Avoids the processing and presentations of antigens and plasma reduce immune complexes. It has shown to inhibit the expression of 2b-3a glycoprotein inactivating platelets, reverses the complex formation in the cellular membrane of β2GPI antibodies and Phospholipids. There's few clinical evidence of its antithrombotic effects in AFAS. The treatment guidelines consider hydroxychloroquine as a potential adjuvant therapy, particularly to its excellent safety profile. New anticoagulant agents: The role of the new anticoagulant agents in AFAS is yet not determined. There are few clinical randomized clinical assays that evaluate its role in the AFAS treatment. Statins: The use of statins could be beneficial in preventing thrombosis, because hypertriglyceridemia and low levels of HDL-Cholesterol are the most frequent cardiovascular risk factor in patients with AFAS. Nevertheless they have also proved to have an antithrombotic and anti-inflammatory effect. Vitamin D has an antithrombotic and immunomodulatory effect through the inhibition of β2GPI antibodies.
In our patient, anticoagulation bridging therapy was started with enoxaparin 60mg SC BID for 5 days and acenocumarol. If AFAS diagnosis is confirmed, the anticoagulation will continue indefinitely with an INR 2-3.
a. Antithrombyn deficiency
b. Protein C or S deficiency
c. Factor V Leiden
d. Double pro coagulant lesion: Prothrombin/Leyden’s V Factor gene mutation
Correct answer: C
The overall prevalence of this primary thrombophilia was described by the Dutch University of Leiden in 1994 as the most common worldwide. It is characterized by a resistance of the Factor V for its inactivation by the protein C and has a current prevalence of approximately 4%, followed by mutation of the prothrombin gene and subsequently high risk thrombophilias, which are characterized by deficient endogenous anticoagulants. Reports in the Mexican population suggest a similar prevalence. Remember that the diagnoses of the 2 most common thrombophilias are based on genetic tests, which yield a high diagnostic certainty [14-16].
a. First-degree relative with unprovoked venous thrombosis
b. Thrombosis of unusual site
c. Multiple Thrombosis
d. All of the above
Correct answer: D.
The evidence of thrombosis in patients diagnosed with primary thrombophilia is scarce, however the most important association in the diagnosis of primary thrombophilia is a history of unprovoked thrombosis in a first-degree relative (mother, father or sibling) or familial tendency of venous thrombosis defined as 2 or more relatives with unprovoked venous thrombosis. The diagnosis of multiple thrombosis has a higher association with the phenomenon of cardio embolism (i.e. Atrial Fibrillation) [17,18]. A tendency of retinal vein thrombosis and the presence mutation of prothrombin gene especially in patients receiving hormonal replacement therapy is seen in primary thrombophilia. Venous thrombosis of unusual site refers to venous thrombosis in retinal, cerebral and intra-abdominal veins. In the case of this patient we have an event of unprovoked thromboembolism in unusual site in a young patient with no risk factors [17,19-21].
a) In the acute event, the coagulation cascade and all pro and anticoagulant events are modified due to thrombosis itself and the effect of anticoagulation therapy
b) Women of reproductive age because of high risk of complications in this group and patients with hematologic double injury pro coagulant (extremely rare prevalence) that could benefit from prolonged or indefinite anticoagulation
c) A y B
d) Patient with a mutation in the prothrombin gene, protein S or C deficiency (considered S high-risk thrombophilia) do not change the choice or duration of treatment
Correct answer: B
Identification and diagnosis of a patient with primary thrombophilia has no utility in the acute event of thrombosis for 2 main reasons: in the acute event, the coagulation cascade and all pro and anticoagulant events are modified due to thrombosis itself and the effect of anticoagulation therapy. The second reason is that identifying a patient with a mutation in the prothrombin gene, protein S or C deficiency (considered s high-risk thrombophilia) does not change the choice or duration of treatment. Two scenarios could probably glimpse benefit: women of reproductive age because of high risk of complications in this group and patients with hematologic double injury pro coagulant (extremely rare prevalence) that could benefit from prolonged or indefinite anticoagulation. Younger patients (<40 years) without comorbidities, have higher probability of a positive pro coagulant profile as in the case of our patient. As mentioned before, the two most common primary thrombophilia’s are diagnosed with genetic tests, whose results are not affected by the acute event of thrombosis and although there is no conclusive evidence of the usefulness of measuring levels of antithrombin, protein S or C in the acute event, theoretically the presence of normal values has a high negative predictive value for primary thrombophilia [20].
a. Yes, Dimer D
b. Yes, serum Thrombin
c. No impact on decision-making regarding treatment
d. Repeat the thrombophilia profile to avoid false positives
Correct answer: C
Thrombophilia profile includes: Antithrombin: functional assay (Anti-Xa cofactor activity). Protein C: functional assay (Chromogenic with snake venom). Protein S: unspecific functional assay, total or free antigen can be measured. Activated Protein C Resistance: based on aPTT with or without Factor V poor plasma. The result must be confirmed with Genotype for Factor V Leiden G20210A mutation. Antiphospholipid antibodies: phospholipid dependent tests for lupus anticoagulant (with Kaolin Clotting Time (KCT) and Russell viper venom diluted time (dRVVT) and anticardiolpin antibodies [22] (Table 3).
Diagnostic tests are of less value than the clinical characteristics of the patient as a predictor of recurrence. Laboratory studies should be limited to the subgroup of patients where results impact treatment (antiphospholipid syndrome, pregnancy, family history or hereditary disease history). Only in some cases the studies must be repeated in optimal conditions to avoid false positives [1]. There are some studies that have attempted to demonstrate the need to restart the anticoagulant therapy in patients who had a previous episode of spontaneous thromboembolism and received treatment for at least 6 months. D-dimer, serum levels of Thrombin and genetic studies are some of them although the cost-benefit evidence is not clear [23].
In this case venous thrombosis occurs in the context of a prolonged aPTT, confirmed 12 weeks after the acute episode with low levels (but not deficiency) of protein C and S in addition to a diagnosis of a hereditary symptomatic erythrocyte membrane disorder. We can conclude that regularly repeated studies on pro coagulant hereditary disease (thrombophilia) is not cost-effective and does not impact the treatment which is based on risk stratification according to clinical characteristics. However it is important to always individualize the case, especially in patients presenting with various bleeding disorders since in these cases it is clear that there is a greater risk of recurrence.
a. 3 to 6 months
b. 1 year
c. Undefined
d. Individualized
Correct answer: D
Taking into account the determinants of the recurrence of a first episode of venous thrombosis help in making treatment decisions. Age (> 65 years), history of a previous episode, antiphospholipid syndrome diagnosis, male and proximal thrombosis are some of them [22]. There are risk stratification models that identify patients at high risk of recurrence where the indefinite anticoagulant therapy is mandatory in contrast to patients with lower risk [24].
The optimal long term management strategy in patients with an episode of venous thromboembolism (VTE) is controversial. The evidence is irrefutable in patients with VTE and cancer as well as in patients with a second episode of unprovoked VTE where treatment duration is undefined [25]. We must take into account the risk of bleeding versus the risk of recurrence. The evidence show that during the first 6 months to 1 year the risk of thromboembolism and major bleeding is the same, however after the first year the risk of VTE recurrence significantly decreases and the risk of major bleeding keeps the same. According to the Current Antithrombotic Therapy and Prevention of Thrombosis Guidelines, a patient with a low or moderate bleeding risk, extended anticoagulant therapy is recommended. The continuing use of treatment should be reassessed annually [26].
In our patient the treatment was indicated for at least 6-12 months. Sex, location and diagnosis of Antiphospholipid Antibody Syndrome are factors to consider in the decision to extend the treatment however we must stratify and take into account the risk of long-term bleeding [27].

![]()

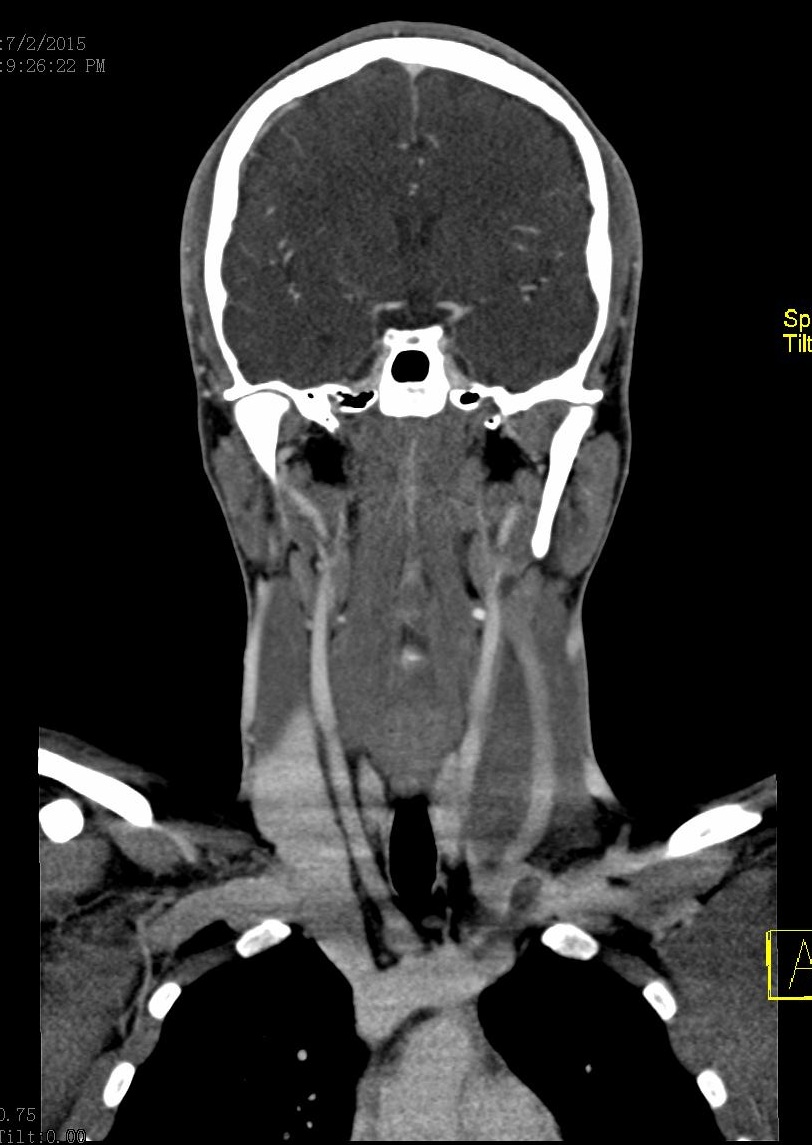 |
Figure 1: Contrast-enhanced CT (venous phase) Axial view: Filling defect is observed at the junction of the middle and lower third of internal left jugular vein with 95% occlusion of venous outflow |
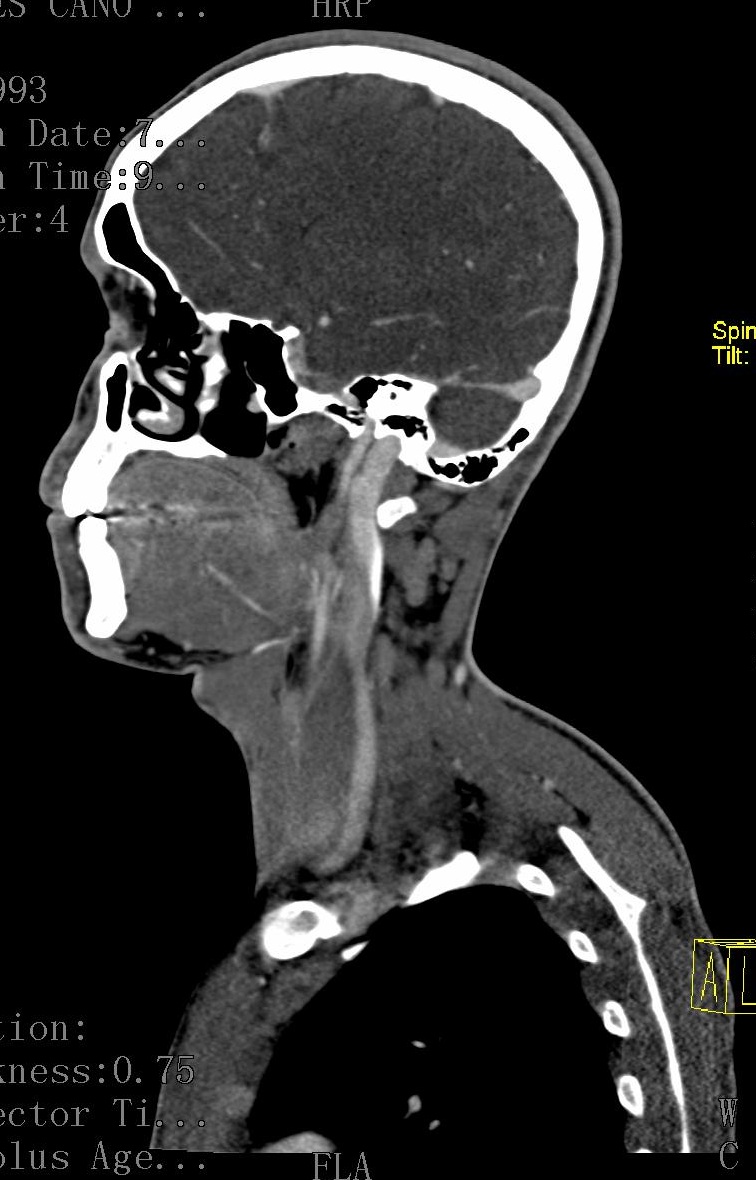 |
Figure 2a: Sagittal section prior administration of contrast: Filling defect is observed at the level of left internal jugular vein |
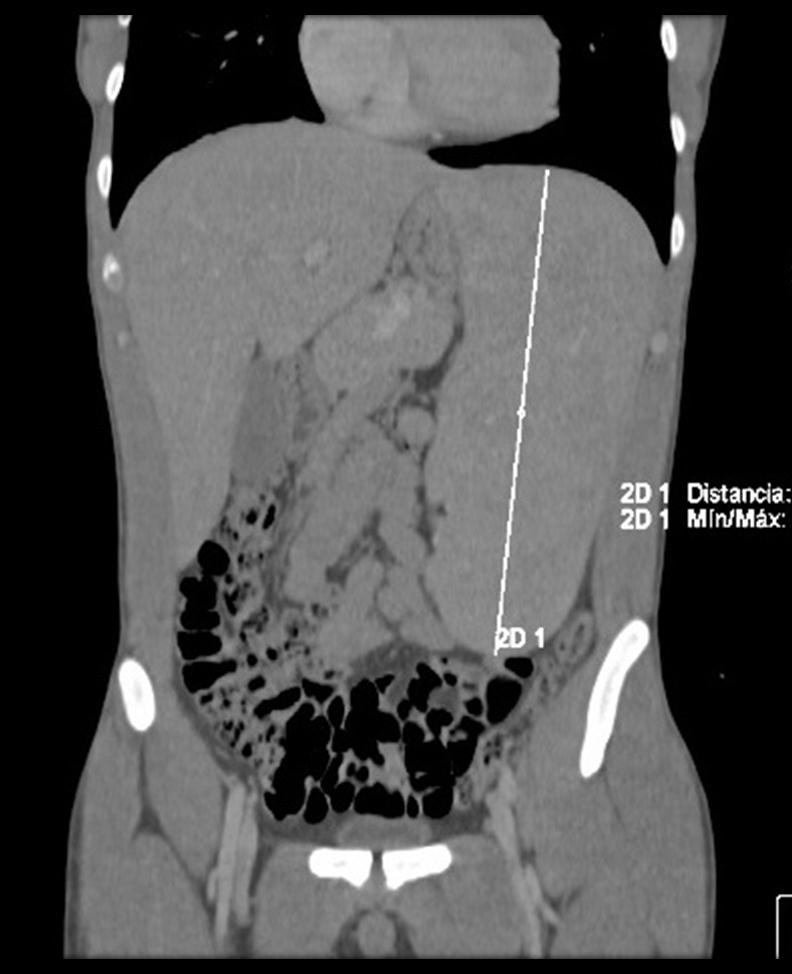 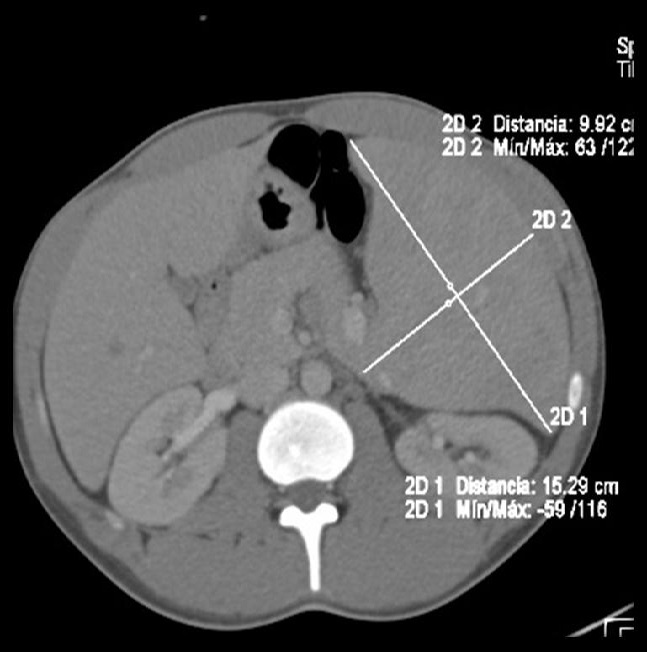 |
Figure 2b and 3: Abdominal CT angiography, coronal and axial view: massive splenomegaly measuring 23.25 x 15.29 x 9.92cm associated with retroperitoneal peripancreatic lymphadenopathy |
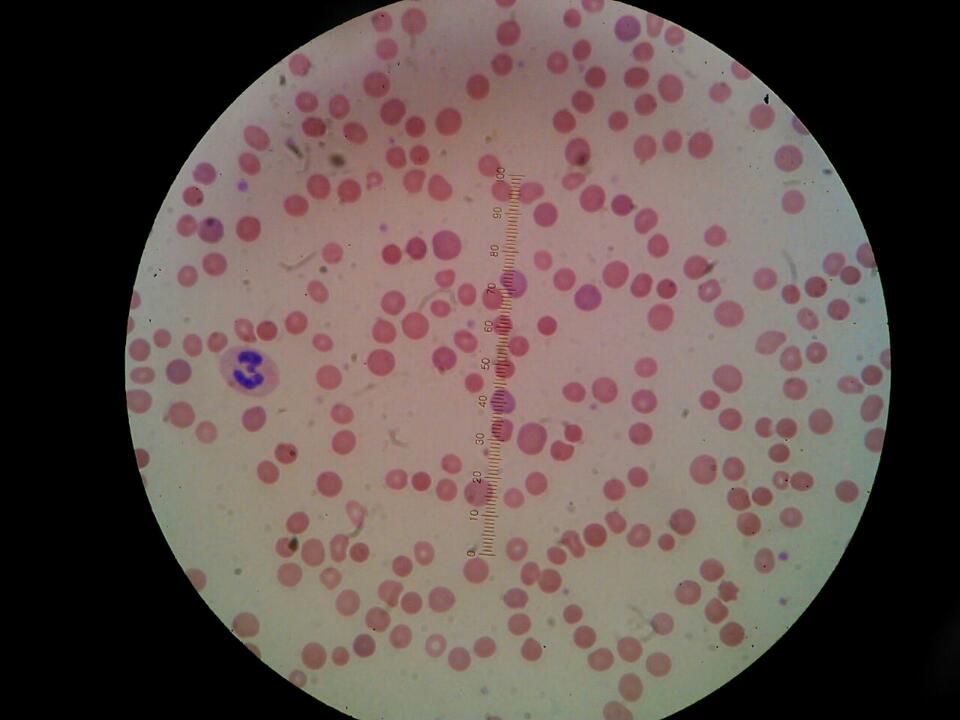  |
Figure 4 and 5: Peripheral blood smear: Erythrocytes lacking of pale center (typical characteristics of spherocytes). Hypochromic microcytic cells with central halo, some with minimum amount of hemoglobin (ghost cells). Macrocytes without polychromasia. Oval-shaped cells with no paralleled halos. Normochromic cytoplasm and hypochromic in others without polychromasia |
Complete Blood count |
Coagulation tests |
||||
|---|---|---|---|---|---|
Parameter |
Result |
Reference range |
Parameter |
Result |
Reference range |
Hemoglobin |
12.3 g/dl |
14-18 g/dl |
TP |
12 secs. |
9-12 secs |
Hematocrit |
34.4 % |
42-54% |
TTP |
48.1 secs. (control 26.7) |
24.4-31.8 secs. |
MCV |
84.5 fL |
80-96fL |
Correction |
- |
|
MCH |
30.1pg |
27-33 pg |
Dilution 1:2 (patient/control) |
37 secs |
- |
MCHM |
25.5% |
11.5-15.5 % |
Dilution 1:4 (patient/control) |
30.5 secs |
- |
RDW |
25.5% |
11.5-15.5 % |
Dilution 1:8 (patient/control) |
27.4 secs |
- |
Platelets |
168 x10 ^3/μL |
150-450 x10 ^3/μL |
INR |
1.09 |
- |
Leucocytes |
9.4 x10 ^3/μL |
4.5-11 x10 ^3/μL |
Dimer D |
390 ng/ml |
0-199 ng/ml |
Neutrophils |
5.6 x10 ^3/μL |
1.8-7 x10 ^3/μL |
|||
Lymphocytes |
3.1 x10 ^3/μL |
1.2-4 x10 ^3/μL |
|||
Reticulocytes |
16% |
<2% |
|||
Haptoglobin |
<5 mg/dl |
- |
|||
Liver Function Tests |
Serum Electrolytes |
||||
Parameter |
Result |
Reference range |
Parameter |
Result |
Reference range |
Total proteins |
7.3 g/dl |
6.1-7.9mg/dl |
Sodium |
139 mmol/L |
136-144 mmol/L |
Albumin |
5.01 g/dl |
3.5-4.8 mg/dl |
Potassium |
3.79 mmol/L |
3.6-5.1 mmol/L |
Globulins |
2.3 g/dl |
2.3-3.8 mg/dl |
Chlorine |
105.2 mmol/L |
101-111 mmol/L |
Total Bilirubin |
4.98 |
0.4-1.5 mg/dl |
CO2 |
27 mmol/L |
22-32 mmol/L |
Direct Bilirubin |
0.25 |
0-0.3 mg/dl |
Corrected Calcium |
8.8 mg/dl |
8.9-10.3 mmol/L |
Indirect Bilirubin |
4.73 |
0.3-1.4 mg/dl |
Phosphorus |
4.7 mg/dl |
2.4-4.7mg/dl |
ALT |
22 U/L |
17-63 U/L |
Magnesium |
1.89 mg/dl |
1.8-2.5mg/dl |
AST |
26 U/L |
15-41 U/L |
|||
AF |
75 U/L |
32-91 U/L |
|||
GGT |
12 U/L |
7-50 U/L |
|||
LDH |
368 U/L |
49-397 U/L |
|||
Blood Chemistry |
Urinalysis |
||||
Parameter |
Result |
Reference range |
Parameter |
Result |
|
Glucose |
95mg/dl |
72-99mg/dl |
Bilirrubin |
1+ |
|
BUN |
11.3 mg/dl |
8-20mg/dl |
Proteins |
traces |
|
Urea |
24.2 mg/dl |
17.1-42.8 mg/dl |
|||
Creatinine |
0.76 mg/dl |
0.44-1.03 mg/dl |
|||
APS panel |
Coagulation Panel |
||||
Parameter |
Result |
Reference range |
Parameter |
Result |
Reference range |
Lupic Anticoagulant |
Detected |
- |
Direct Coombs |
Positive |
Negative |
Ac IgA anticardiolipin |
<9 |
- |
Protein S activity |
48% |
70-150% |
Ac IgG anticardiolipin |
>560 |
>80 positive |
Protein S antigen |
80% |
60-150% |
Ac IgM anticardiolipin |
<2 |
0-12.9 |
Total Protein S |
80% |
58-150% |
Anti beta 2 glycoprotein |
1.2 |
- |
Free Protein S |
61% |
56-124% |
Protein C activity |
57% |
70-180% |
|||
Protein C antigen |
62% |
70-140% |
|||
Mutation 20210 |
Normal |
Normal |
|||
Leiden mutation |
Normal |
Normal |
|||
Viral Panel |
|||||
Parameter |
Result |
Reference range |
Parameter |
Result |
Reference range |
HBV HCV |
Non-reactive |
Non-reactive |
Hip osmotic solution |
||
HIV 1 y 2 |
Non-reactive |
Non-reactive |
0.85% (initial hemolysis) |
Positive |
0.45-0.5% |
VDRL |
Positive 1:8 |
Negative |
0.55% (complete hemolysis) |
Positive |
0.3-0.4% |
Table 1: Laboratory Tests |
|||||
Diagnosis recommendations in HS [2] |
|---|
1. Newly diagnosed patients with a family history of HS, typical clinical features (splenomegaly) and laboratory investigations (spherocytes, raised MCHC, increase in reticulocytes) do not require any additional tests (1A). |
2. If the diagnosis is equivocal, e.g. where there are a few spherocytes on the film but no other laboratory, clinical or family evidence, a screening test with high predictive value for HS is helpful. The recommended screening tests are the cryohemolysis test and EMA binding (1A). |
3. Confirmation of diagnosis may be necessary in selected cases if the screening tests produce an equivocal or borderline result. Gel electrophoresis analysis of erythrocyte membranes is the method of choice. |
4. Diagnosis of HS does not require further investigation by molecular analysis of the affected genes |
Thrombophilias most common primary prevalence and relative risk in clinical manifestations [28] |
Antithrombin deficiency |
Protein C deficiency |
Protein S deficiency |
Factor V Leiden |
Prothrombin 20210A mutation |
Lupus anticoagulant* |
Anti-cardiolipin antibodies |
Anti-B2 GPI antibodies |
|
|---|---|---|---|---|---|---|---|---|
Prevalence in the general population |
0.02% |
0.2% |
0.03%-0.13% |
3-7% |
0.7-4% |
1-8% |
5% |
3.4% |
Relative risk for a first venous thrombosis |
5-10 |
4-6.5 |
1-10 |
3-5 |
2-3 |
3-10 |
0.7 |
2.4 |
Relative risk for recurrent thrombosis |
1.9-2.6 |
1.4-1.8 |
1.0-1.4 |
1.4 |
1.4 |
2-6 |
1-6 |
No data |
Relative risk for arterial thrombosis |
No association |
No consistent association |
No consistent association |
1.3 |
0.9 |
10 |
1.5-10 |
No data |
Table 3: Prevalence of thrombophilia and relative risk estimates for various clinical manifestations [28] |
||||||||




































