Open Access
Research Article
Max Screen >>
ISSN: 2455-7641
Copyright: © 2016 Liljeholm M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Background: Congenital dyserythropoietic anemia type III (CDA III) can be caused by mutation in KIF23. CDA III differs from CDA I and II in the sense that secondary hemochromatosis has not been reported. However, we have observed elevated serum ferritin in a CDA III family. Since primary hemochromatosis is common in Northern Europe we decided to screen the family for HFE mutations.
Aim:Study clinical appearance and prevalence of HFE gene mutations, C282Y and H63D, in a CDA III family.
Methods: DNA from 37 CDA III patients and 21 non-affected siblings was genotyped. Iron status from EDTA plasma was measured in 32 of the CDA III patients and 18 of the non-affected siblings.
Results: Out of 37 CDA III patients, 18 carried heterozygous HFE mutations and six were compound heterozygotes. Out of 21 CDA III negative siblings, nine had heterozygous HFE mutations, two were homozygous (one H63D and one C282Y), and two were compound heterozygous. None of the patients with wt HFE, regardless of CDA III status, suffered from iron overload. Four patients with HFE mutations needed treatment with phlebotomy to normalize ferritin and transferrin iron saturation; one CDA III negative patient with homozygous C282Y, two CDA III patients with heterozygous HFE mutations and one CDA III case with compound heterozygosity.
Conclusion: HFE mutations were found in 65 % of CDA III patients and in 62 % of their CDA III negative siblings. Heterozygous HFE mutation, C282Y and even H63D, can cause iron overload when occurring concomitantly with CDA III.
Keywords: Congenital dyserythropoietic anemia; Hereditary hemochromatosis; Iron overload; HFE gene; KIF23 gene
Congenital dyserythropoietic anemias (CDA) is a group of rare hereditary hemolytic disorders. Three major subtypes (I, II, III) and some minor entities (IV-VII) have been described [1]. The CDAs present with laboratory signs of increased hemoglobin (Hb) turnover, such as low or absent haptoglobin, elevated lactate dehydrogenase (LDH) and bilirubinemia. The bone marrow is hypercellular with a predominance of large bi- to multi-nucleated erythroblasts. Erythroblast morphology differs between CDAs, but the diagnosis of CDA I, II and III is best set by genetic testing [2,3].
CDA I and II are the most common forms, both with autosomal recessive inheritance [4-6]. Symptoms and signs of CDA I and II are mainly mild to moderate anemia, splenomegaly, and iron overload. The risk of secondary hemochromatosis increases when transfusions are needed [1]. Overexpression of Growth Differentiation Factor 15 (GDF15) in CDA I and II has, as in thalassemia, been shown to limit hepcidin expression thereby increasing iron absorption [7,8].
CDA III is the rarest form of the major CDA subtypes with only about 60 reported cases worldwide. A familial form with dominant inheritance has been described in two families, one American family with four cases and one family in Västerbotten County, Sweden, with the majority of reported cases [9,10]. One family in Argentina with eight cases has been described, although the pattern of inheritance in this family has not been confirmed [10]. A few cases of CDA III with recessive inheritance have been described in the literature [11,12]. One of these patients developed iron overload after erythrocyte transfusions [12].
In the Västerbotten CDA III family, the disease is caused by a mutation in c.2747C>G, p.P916R in KIF23. The mutation results in a dysfunctional KIF23 protein, also known as Mitotic Kinesin Like Protein 1 (MKLP1), leading to failure of cytokinesis, the final stage of mitosis. As a result, the bone marrow in CDA III is characterized by gigantoblasts, large erythroblasts with up to 12 nuclei in the same cell [13].
The Swedish CDA III family has been mapped through six generations back to the middle of the 19th century [14,15]. The disease presents with mild to moderate anemia, rarely in the need of red cell transfusions. Hemolysis is extra- as well as intravascular, leading to hemosiderinuria. Since CDA III is a disorder of ineffective erythropoiesis, hepcidin is expected to be suppressed, leading to iron overload, but ferritin levels were not found to be elevated in CDA III patients in earlier studies [15]. Intravascular hemolysis with hemosiderinuria and loss of iron in the urine has been suggested to protect patients against iron overload [15].
However, we have recently observed elevated ferritin levels in some CDA III positive and negative members of the Västerbotten family. Further investigations revealed mutations of the hemochromatosis gene (HFE), the gene involved in the development of hereditary hemochromatosis (HH). HFE mutations (C282Y and H63D) are frequently found in Northern Europe and the prevalence of HH in Sweden is 0.5% [16]. Homozygous mutation of C282Y or compound heterozygosity of C282Y and H63D reduces hepcidin, thereby increasing iron absorption, and may result in clinical signs of hemochromatosis, but the penetrance is relatively low, estimated in the literature to 10-30% [17]. Heterozygous C282Y is found in 7% of the Swedish population and 0.5% carry the mutation on both alleles [16]. Heterozygous H63D is frequently found all over the world with a prevalence of about 10-20% but does not by itself lead to significant iron overload [17,18]. Treatment of iron overload in patients with primary hemochromatosis is based on phlebotomy.
We have studied the clinical appearance and presence of mutations in KIF23 and HFE in the Västerbotten CDA III family.
All patients belonged to the “Västerbotten CDA III family” (Figure 1). None of the participants in the study was transfused.
DNA was available from 37 individuals with CDA III and 21 non-affected siblings. The status of CDA III was confirmed by detection of KIF23 c.2747C>G as earlier described [13].
Sequencing of KIF23 was performed by polymerase chain reaction (PCR). The products of sequencing reactions were analysed on ABI 3500xL Dx Genetic Analyzer (Applied Biosystems, Foster City, CA). PCR - products were digested by endonuclease, separated by electrophoresis on an Agarose gel and visualized after staining with ethidium bromide [13].
HFE analysis was done with TaqMan® SNP Genotyping Analysis. Assay for detecting c.187C>G, p.H63D, rs1799945 was done using predesigned assay C1085600_10 (Applied Biosystems). Assay used for detecting c.845G>A, p.C282Y, rs1800562 was designed using Custom TaqMan® Assay Design Tool.
Hepcidin was analysed using a commercially available Competetive ELISA test (Bachem, Peninsula Laboratories, LLC, CA, USA).
Blood samples in EDTA-tubes were used for hematological analyses including iron status (ferritin and TSAT) and hematological parameters (Hb and LDH). Reference intervals for ferritin were 30-400 ug/L for males and 13-150 ug/L for females. Upper limit of normal for TSAT was < 60% for males and < 50% for females. From 1997 to 2007 ferritin was analysed by Boehringer Mannheims Tina-Quant kit 1661400. From 2007 analyses were done using Roche Modular E170 with electrochemiluminescence immunoassay (ECLIA). Correlation between methods was 1.0. Serum-Fe was analysed with Hitachi 717/Hitachi 911, Boehringer Mannheims Ferrozine 1997-2007 and thereafter with Hitachi 911, delta, till Vitros 5.1 FS. Transferrin analysed with Vitros 5,1. TSAT was calculated by the following formula: S-Fe [μmol/L] x 100 / (S-Transferrin [g/L] x 25,1).
All individuals with HFE mutation and elevated TSAT were offered clinical and laboratory investigation to confirm or reject iron overload. Phlebotomy was initiated in individuals with iron overload and in patients with homozygous C282Y plus TSAT above the upper limit of normal.
Ferritin, TSAT, Hb and LDH are presented as median values and range. Two-sided Mann-Whitney U-test was used for comparison between the CDA III positive and negative group. P < 0.05 was regarded as significant.
Ethical approval was obtained from the Regional Ethics Committee in Umeå, Sweden, and informed consent was obtained from all patients.
The group of patients with CDA III and available iron status and hematological parameters (Hb and/or LDH), consisted of 16 males and 16 females with a median age of 53 years, and the CDA III negative group consisted of 10 males and 8 females, with a median age of 44 years. The median hemoglobin value among CDA III patients was 116 g/L (94-157), LDH 6.2 ukat/L (4.2-10), and bilirubin 18 umol/L (7-86).
Ferritin was higher (108 umol/L) in the CDA III positive group than in the CDA III negative group (76 umol/L), and TSAT was also higher among CDA III patients, 46% vs. 34%, but the differences were not significant (p=0.055 for both comparisons). The two groups did not differ concerning age or gender.
HFE genotyping was successfully performed in all 58 cases. HFE mutation was found in 24 out of 37 CDA III patients (65%) and 13 out of 21 CDA III negative siblings (62%), (Figure 1). Among the 50 cases with available iron status, 21 out of 32 CDA III positive (66%) and 11 out of 18 CDA III negative siblings (61%) were found to have HFE mutations. 16 of the CDA III patients with HFE-mutation were heterozygous either for C282Y or H63D, and five were compound heterozygotes. Seven of the 18 CDA III negative siblings were heterozygous carriers of HFE mutations, two were compound heterozygotes, and two had homozygous HFE-mutations (Table 1). In individuals with heterozygous C282Y (9 CDA III positive and 6 CDA III negative cases), ferritin levels were significantly higher in CDA III patients, p=0.036.
Laboratory evidence of iron overload was noted in four patients, one CDA III negative case with homozygous C282Y, and three CDA III positive individuals with heterozygous HFE mutations (Table 2). Magnetic resonance imaging (MRI) of the liver and S-hepcidin analysis was performed only in the CDA III positive patient with heterozygous H63D mutation. Liver iron was 6700 ug/g dry tissue (normal range 300 -1500) and hepcidin was 46 ug/L (normal range 8-76). To normalize ferritin and TSAT, treatment with phlebotomy was started in four patients; the CDA III negative sibling who was homozygous for C282Y and three CDA III patients, one compound heterozygote and two with heterozygous mutations, one C282Y, one H63D.
A total of 18 individuals, 11 CDA III cases and 7 CDA III negative siblings, had wt HFE. Ferritin levels did not differ between these groups (p=0.86, Table 1). One of these CDA III positive patients, suffering from hypertension and hyperlipidemia, had elevated ferritin (642 ug/L) but TSAT was not elevated, 40%. In another CDA III positive case without HFE gene mutation TSAT was elevated (73%) due to subnormal transferrin 1.68 (reference interval 1.87-3.19 g/L). Ferritin was not elevated in this patient, 191 ug/L.
We found that 65% of CDA III patients and 62% of their CDA III negative siblings in the Västerbotten family carried mutations in the HFE gene. Two CDA III patients with heterozygous HFE-mutations and one CDA III patient who was compound heterozygous for the HFE- mutations needed treatment with phlebotomy due to iron overload.
One of the heterozygous cases carried H63D, which is not reported to lead to iron overload by itself. MRI of the liver confirmed iron overload in this patient. Hepcidin was in the normal range, but we believe that the level was inappropriate considering the iron status, indicating disruption of the feedback system. We hypothesise that increased erythropoiesis produces larger amounts of erythroferrone suppressing hepcidin that may be further suppressed by HFE mutation.
None of the healthy siblings carrying HFE-mutations with heterozygous or compound heterozygosity nor CDA III positive cases with wt HFE had laboratory signs of iron overload. One CDA III positive individual with wt HFE had elevated ferritin and one had elevated TSAT. After further investigation none of these patients fulfilled the criteria of iron overload. Ferritin levels were higher among CDA III positive vs CDA III negative individuals with heterozygous C282Y, though study population is limited. Earlier studies have shown that homozygous mutation of HFE C282Y or, eventually, compound heterozygosity of C282Y and H63D is needed to develop clinical signs of hemochromatosis in healthy individuals, though penetrance is relatively low [17]. In this study we found that heterozygous mutation of the HFE- gene, either C282Y or H63D can be sufficient to cause iron overload in patients with CDA III. All patients were screened for diabetes since this is a well-known metabolic factor causing elevated ferritin, although TSAT is not elevated in diabetes [19]. One male CDA III patient, heterozygous for H63D, was diagnosed with type II diabetes mellitus. Except for elevated ferritin (1084 ug/L), TSAT was also elevated (80%) in this patient, indicating iron overload.
Earlier studies on HFE-mutations in patients with hematologic disorders where iron overload can be a problem, such as thalassemia and myelodysplastic syndromes (MDS), are contradictory. Some studies show iron overload in thalassemia patients with co-existing HFE-mutations, even in heterozygous forms [20-22]. Recently, non-transfused MDS-patients, carriers of HFE-mutations, were found to have higher ferritin level and inferior overall survival compared to non-mutated MDS-patients [23]. However, other studies have not verified that heterozygous mutation affects iron overload in thalassemia [24,25].
So far, phlebotomies have been performed without problems in spite of the continuous hemolysis in our CDA III patients and phlebotomy alone is sufficient to control ferritin and TSAT-levels.
This study indicates that heterozygous HFE mutation, C282Y or H63D, is sufficient to cause pathologic iron overload when occurring concomitantly with CDA III. Therefore, in order to prevent organ damage in these patients, screening for primary hemochromatosis should be performed in all patients with congenital dyserythropoietic anemia type III.
Financial support was obtained through regional agreement between Umeå University and Västerbotten County Council on cooperation in the field of Medicine, Odontology and Health (ALF), and unrestricted grants from Cancerforskningsfonden Norrland. None of the authors has any conflict of interest to declare.

![]()

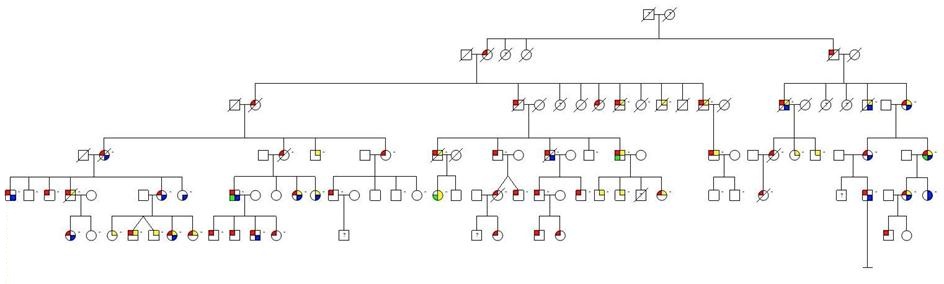  |
| Figure 1: Pedigree of the Västerbotten CDA III family. Participants with available DNA are indicated with dash (–) to the right of the individual symbol |
HFE status |
Ferritin ug/L CDA III + CDA III – |
TSAT % CDA III + CDA III – |
Hb g/L CDA III + CDA III – |
LDH ukat/L CDA III + CDA III – |
||||
|---|---|---|---|---|---|---|---|---|
HFE wt |
116 (28-642)* n=11 |
90 (28-392) n=7 |
45 (26-73) ** n=11 |
32 *** |
119 (105-135) n=11 |
135 (118-153) n=7 |
6.1 (4.2-8.0) n=11 |
2.8 (1.9-4.5) n=7 |
C282Y +/- |
89 |
73 (13-154) n=6 |
45 (37-100) n=9 |
33 (21-46) n=6 |
123 (107-157) n=9 |
145 (114-154) n=6 |
6.0*** (5.2-10.0) n=9 |
2.8 (1.9-3.1) n=6 |
C282Y +/+ |
229 n=1 |
79 n=1 |
153 n=1 |
2.1 n=1 |
||||
C282Y +/- H63D +/- |
66 |
75 |
55 |
49 |
116 |
154 |
6.4*** |
2.4 |
H63D +/- |
139 |
3
|
39 |
4
|
105 |
94 |
6.7*** |
2.4 |
H63D +/+ |
20 n=1 |
49 n=1 |
138 n=1 |
2.7 n=1 |
||||
| +/- heterozygous mutation, +/+ homozygous mutation * elevated ferritin in one patient with hyperlipidemia and normal TSAT ** elevated TSAT in one patient with subnormal Tf and normal ferritin *** sample missing in one case Table 1: Iron status, Hb and LDH in individuals of the Västerbotten CDA III family, with and without HFE mutation. Medians and (range) |
||||||||
| CDA status | Gender and age (years) | HFE status | Ferritin (ug/L) | TSAT (%) | Hb (g/L) | LDH (ukat/L) |
|---|---|---|---|---|---|---|
| wt | F 49 | C282Y +/+ | 229 | 79 | 153 | 2.1 |
| CDA III + | M 60 | C282Y +/- | 987 | 100 | 143 | 5.2 |
| CDA III + | F 61 | C282Y +/- H63D +- |
700 | 55 | 111 | 6.1 |
| CDA III + | M 60 | H63D +/- | 1084 | 80 | 135 | 6.3 |
| Table 2: Individuals in whom phlebotomy has been initiated due to iron overload | ||||||




































