Open Access
research article
Max Screen >>
ISSN: 2455-7625
Copyright: © 2014 Capobianco E. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Recently, the pathogenesis of coronary atherosclerosis in a swine model has been investigated by considering in particular the effects of a high cholesterol diet lasting 8 or 16 weeks [1]. The proposed systems medicine approach integrating data of plasma adhesion molecules, cytokines, lipoproteins, tissue proteins and histology allowed for an assessment of their relationships with artery-specific proteins. We send the reader to the seminal paper for experimental and computational details, and also data collection modalities. While macrophage-related proteins were found significantly and positively associated to the atherogenesis-inflammatory disorder, the study of protein connectivity patterns remains to be investigated. This is also a relevant step, because such type of evidence synthesis could lead to signatures of markers. Through the popular tool STRING [2], we aim to inspect protein-protein interaction networks (PIN), i.e. whether they can generate interpretable and testable hypotheses in such a complex experimental context with potential clinical impact.
From the available coronary secreted protein data (n=200) obtained by LC- MS identifications and peptide peak related expression values, the PIN configurations are obtained in two steps. First, by looking at sample-specific protein profiles, and by selecting the most significant values (thresholding step). Second, moving beyond the role assigned to proteins of potential mediators or markers of local inflammation underlying coronary atherosclerosis lesions, the identification of motifs or patterns in PIN could be valuable to predict the effects of diet regimens (mapping step). The advantage of network inference over classical statistical models is primarily referred to the benefit of avoiding problems of unbalanced data, by which disproportionate amounts of samples and variables are present in the model. The main consequence of this imbalance of data is that subset selection for training, testing and validation is not ensuring good classification performance, for instance, and regression models require some regularization in order to produce consistent results. The limitation of protein- centric network analysis comes from the fact that other datasets are not taken into consideration. Naturally enough, if redundancy is present, the analysis does not suffer from any power loss. A way to go is to exploit the biological annotations associated to the protein networks. In our context, with two main groups of samples subject to different treatment conditions, the expectation is that PIN features may appear as specifically characterizing such groups, and such characteristics may translate into some distinctive biological annotations, say biological processes, pathways etc.
Considering only the significantly expressed proteins measured along the sample profiles, and those analyzed under mild (2 months) and severe (4 months) diet conditions, we observed that ribosomal and mitochondrial protein interactions appeared in a couple of profiles taken from the first diet condition, the mild one, while two sample profiles from the second diet condition, the sever one, provided an interesting common feature. This evidence depends on the criterions by which we have selected the significant expression values of the protein profiles. We have proceeded empirically, by building barplots of expression values for each sample and choosing values outside variability bands. Each profile has an associated PIN, which is then analyzed through STRING in either confidence or evidence modalities.
As the former indicates the strength of the connectivity, only high confidence scores (0.7) attached to the observed protein interactions were retained. The interesting feature that was revealed refers to a protein, AMBP, i.e. a complex glycoprotein secreted in plasma (the alpha-1- microglobulin/bikunin precursor belongs to the superfamily of lipocalin transport proteins). This protein is known to protect cells and tissues from the damage induced by oxidative stress, and may be regulative of the inflammatory processes, even if there is not yet evidence of an established role as a marker or risk factor for cardiovascular diseases [3]. When looking at the AMBP interactions, for one of the two profiles the high confidence interactor that is observed is AHSG (Alpha 2-HS glycoprotein), which prevents glucose toxicity associated with cardiomyocyte dysfunction, and is known to play a role in vascular calcification and plaque development [4], while in the other profile the interactor is the APOC3 protein (Apolipoprotein (apo) C-III), i.e. a small exchangeable apolipoprotein, and major constituent of plasma VLDL and HDL which inhibits lipoprotein lipase and hepatic lipase, thus delaying the catabolism of triglyceride-rich particles. Notably, elevated plasma APOC3 levels are positively correlated with plasma triacylglycerol (TAG) concentration in hypertriglyceridemia subjects. Evidence has also suggested that the deficiency of APOC3 could also exert an effect on cardioprotection [5].
More associations can be described by relaxing the selection of expression values from the sample protein profiles, but the stringency here adopted is sufficient to demonstrate the informativeness of PIN with regards to discriminating between conditions. In other words, differential network configuration analysis is used as an inference strategy, and Figure 1 and 2 show the PIN configurations drawn from both sample profiles. The entire protein expression values are represented, and red circles are used to indicate the proteins that were found significant, i.e. the PIN hotspots. Note also that by exploring the neighbor proteins of the PIN hotspots from both sample profiles, direct interactors are SERPINA 1[6], which helps the control of several types of chemical reactions by blocking (inhibiting) the activity of certain enzymes, and APOA1 (i.e. the most abundant protein of HDL), which circulates in the bloodstream and extract cholesterol from body tissues and transport it to the liver for excretion or recycling. Increased levels of HDL have been correlated with a decreased risk of atherosclerosis---a primary cause of cardiovascular disease. Both these proteins have been widely studied in association with atherosclerosis, while also MFGE 8 (Milk fat globule–EGF factor 8) appears as a first-neighbor indirect interactor, and is known to play a protective role in arteries and to facilitate angiogenesis. These are proteins which have not been expressed at the most significant levels, but if seen in the network context of the identified hotspots, they allow to infer more about the interaction dynamics.
Networks have gained popularity to the point that they are now pervasively applied in systems biology; while a less consolidated role is played in clinical biomedicine. Our study suggests that this potential is worth much consideration in the latter field. First, networks are truly integrative inference tools, displaying relationships between heterogeneous entities. Second, the interactive associations can be measured and assessed in terms of confidence. Third, the analysis turns out to be quite robust: in our application context – despite a very limited number of samples, analysis restricted to just the most significant protein expression values, and consideration of high confidence scores for the interactions- we achieve in a few examples a discriminatory power in selecting protein-protein interactions linked to effects of severe vs mild diet conditions.

![]()

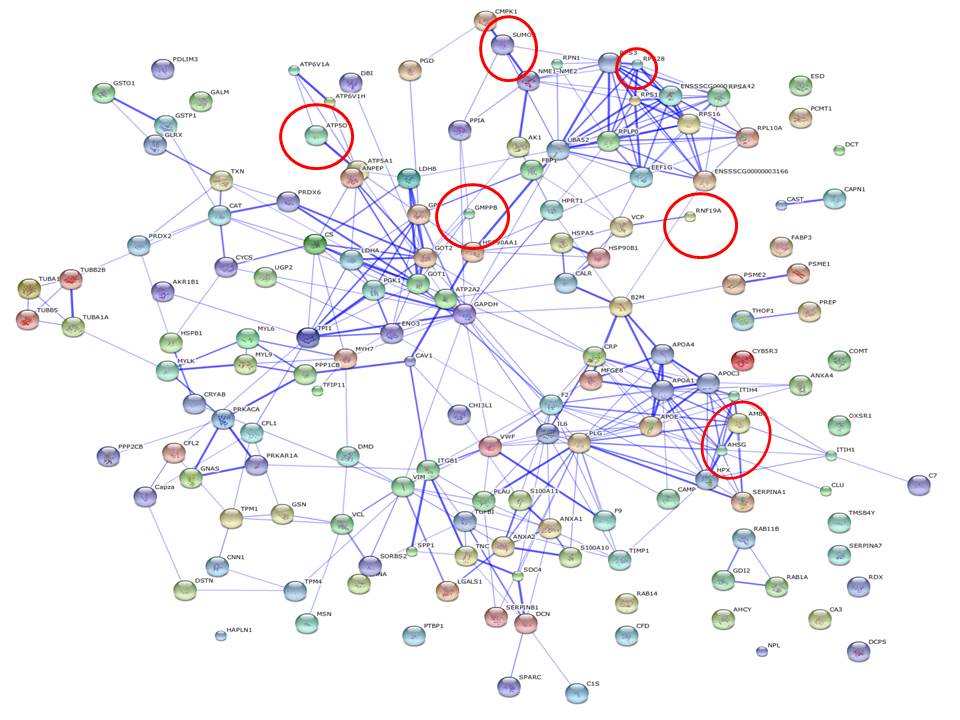 |
| Figure 1: Profile of one of the 6 samples under severe diet condition. |
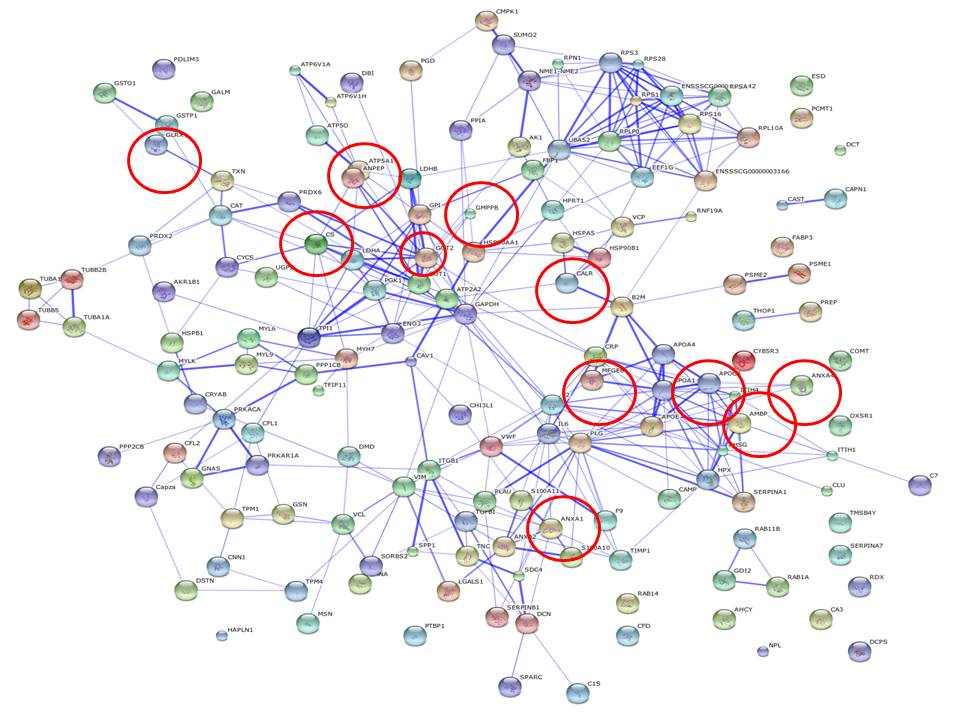 |
| Figure 2: Profile of another of the 6 samples under severe diet condition. |




































