Open Access
Case report
Max Screen >>
ISSN: 2348-9820
Copyright: © 2021 Dou T. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Idiopathic pulmonary hemosiderosis (IPH) extremely rarely occurrs in adults according to current literature.We report one case of IPH in a middle-aged woman presenting with extensive central-predominant ground glass opacities and consolidation in the lungs. Since these imaging findings are not specific and can be similar to other diffuse pulmonary diseases, it is essential to have an deep understanding of this disorder. Imaging findings must be combined with clinical laboratory examination to make a diagnosis of IPH. Transbronchial lung biopsy (TBLB) is the gold standard for evaluation of suspected patients. We review the current literature and provide a recommendation based on discussion.
Keywords:Idiopathic Pulmonary Hemosiderosis; Computed Tomography; Imaging Findings
Idiopathic pulmonary hemosiderosis (IPH) is a rare disorder characterized by recurrent episodes of unexplained intra-alveolar hemorrhage, followed by accumulation of hemosiderin in macrophages. In selected populations, the prevalence of IPH alone is estimated to be 0.24 to 1.23 per [1]. The imaging findings can be similar to infectious leasions on computed tomography (CT). To deepen the understanding of this disorder, we report one case of IPH in an adult confirmed by transbronchial lung biopsy (TBLB) and review relevant literature.
A 47-year-old woman had a 3-month history of cough, sputum, and hemoptysis symptoms, which occurred 3 to 4 times per day with a blood loss of 5 ml each time. She was treated empirically with broad-spectrum antibiotics after being suspected of a nonspecific infection. However, during treatment, symptoms of progressive dyspnea were accompanied. Laboratory tests indicated erythrocytopenia, with a red blood cell count of 3.16x1012/L (normal range, 3.5-9.5x109/L), a white blood cell count of 14.72x109/L (normal range, 3.8-5.1x1012/L), with a decreased hemoglobin level of 73g/L (normal range,115-150g/L). Serum biomarkers for vasculitides and autoimmunity, such as an anti-nuclear antibody, anti-neutrophil cytoplasmic antibody, and rheumatoid factor, were normal. Transbronchial lung biopsy revealed hemosiderin-laden macrophages in alveoli. During treatment, non-enhanced CT of the chest was performed several times.
In the hilar, perihilar, and lower lobe regions, non-enhanced axial and coronal chest CT images revealed significant central ground-glass opacities and consolidation. The lung apices and the costophrenic sulci were spared (Figures 1a and b). After three days of antibiotic treatment, the lesions had progressed significantly (Figures 2a and b). Based on the patient's symptoms, signs, and laboratory tests, idiopathic pulmonary hemosiderosis (IPH) was diagnosed, and the patient was given hormone and immunosuppressive therapy. The lesions were visibly absorbed after one week of treatment and vanished after two weeks (Figures 3a and b).
IPH is most prevalent in children under the age of 10, although it is uncommon in adults, with a mean survival rate of approximately 2.5 years after diagnosis [2].The most common clinical manifestations are chronic pulmonary symptoms (cough, dyspnea, or hemoptysis), iron deficiency anemia, and pulmonary infiltration [3]. However, one of the typical triads may be the leading symptoms.
The definite cause of IPH is still unknown, although, several hypotheses have been proposed for its explanation, including autoimmune, environmental, allergic, and genetic factors [3]. According to one study, patients with IPH are often linked with particular autoimmune illnesses, such as celiac disease, dermatitis herpetiformis, glomerulonephritis, and rheumatoid arthritis [4]. Based on its pathology, IPH is classified into three phases: acute phase, chronic phase, and sequela phase. In the acute phase, a large number of red blood cells are manifested and exudates in alveoli, alveolar edema, and alveolar septum thickening. In the chronic phase, the major symptoms include a large amount of hemosiderin being deposited in the alveolar interstitium, alveolar interstitial fibrous tissue proliferation, interlobular septum, and alveolar wall thickening. The development of extensive interstitial fibrosis within the lung is the most common symptom of the sequela phase. The pathogenic alterations influence the imaging findings. In the acute phase, the chest CT scan image shows extensive ground-glass opacities, which are mostly symmetrically distributed in the pulmonary hilum, middle, and lower lung regions; with the lung apices and costophrenic sulci being less affected. Ground-glass opacities indicate diffuse intra-alveolar hemorrhage when coexistence with consolidation indicates that the alveoli are full of blood. The interlobular septum can thicken due to the deposition of hemosiderin-containing macrophages in the interstitium [5]. The "crazy-paving" sign appears due to interlobular septum thickening coexistence with ground-glass opacities [6]. Recurrent pulmonary hemorrhage patients may develop pulmonary fibrosis, which includes thickening of the interlobular septum, reticulation, and small cystic foci. HRCT is an important tool for detecting diffuse tiny reticulation, fine nodules, and interlobular septum thickening. The patient studied in this report was in the acute phase.
Because of the non-specific imaging results of IPH, it must be combined with a clinical laboratory examination to distinguish it from other diffuse pulmonary diseases. For instance, pulmonary alveolar proteinosis, anti-neutrophil cytoplasmic autoantibodies (ANCA) are associated with systemic vasculitis and phylactic pneumonia. Ground-glass opacity, crazy-paving sign, and map-like alteration are all symptoms of IPH, although they're more common in pulmonary alveolar proteinosis [7]. Furthermore, the lesions of pulmonary alveolar proteinosis often involve the subpleural areas. In ANCA-associated systemic vasculitis, the serum ANCA is mostly positive and multiple organs can be involved, with the kidneys being the most frequently affected organs. The typical CT scan of phylactic pneumonia shows diffuse central lobular nodules, patchy attenuation or ground-glass opacities, and mosaic signs dominated in the upper and middle lobes [8]. It can be distinguished from IPH if the patient with a history of allergen exposure and the lymphocyte percentage in bronchoalveolar lavage fluid (BALF) solution is increased.
IPH is an exclusive diagnosis. The presence of hemosiderin-containing phagocytic cells in sputum, BALF, or gastric aspiration liquid should be verified in patients suspected of IPH [9]. Other diseases such as infectious, cardiovascular, or immune-related diseases should be excluded in patients with pulmonary hemorrhage [10]. TBLB is still the gold standard for the diagnosis of IPH.
IPH is typically treated with medications (glucocorticoid and immunosuppressant). Studies have shown that early administration of glucocorticoid can effectively delay pulmonary fibrosis [10]. In this study, hemoptysis recurred following glucocorticoid decrease, indicating that glucocorticoids should be lowered with caution during IPH therapy.
IPH is rare among adults, and imaging results on single chest CT are not specific. The longitudinal follow-up would be very extremely beneficial in the diagnosis and assessment of efficacy. Early standardized therapy is essential; which would lead to a better outcome, fewer complications, and reduced sequela.
None
The authors claim that none of the material in the paper has been published or is under consideration for publication elsewhere.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.

![]()

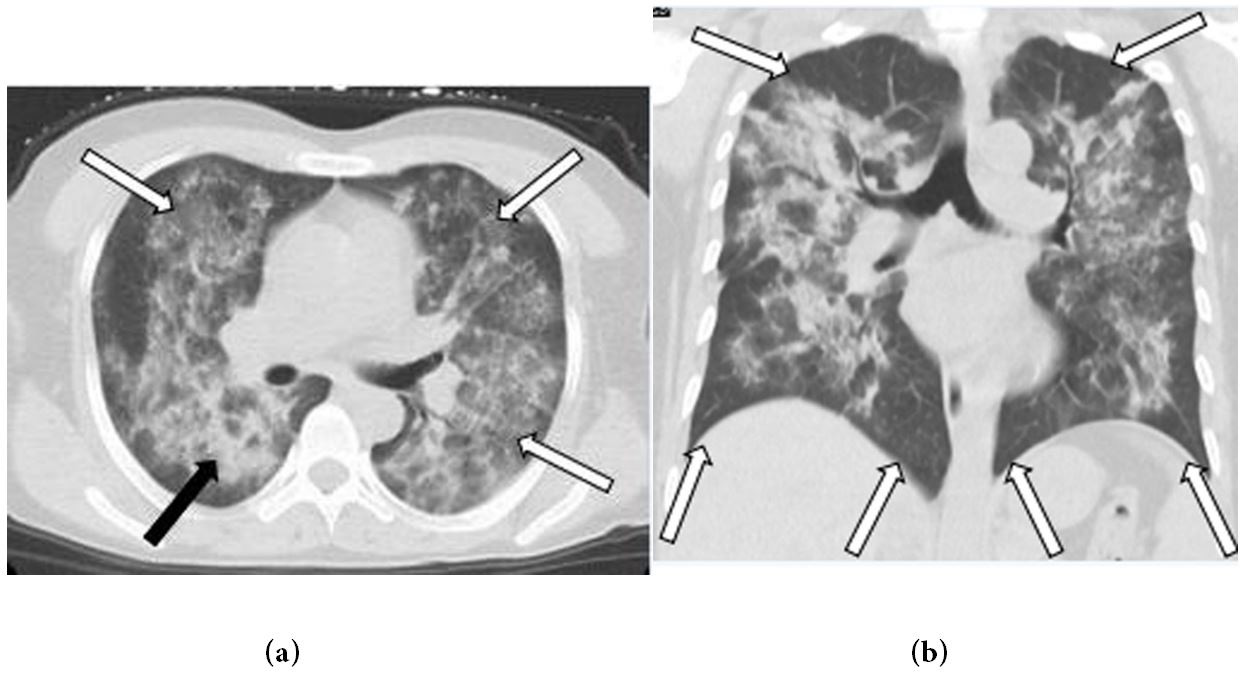 |
| Figure 1: (a) Axial and; (b) coronal non-contrast computed tomography (CT) images of the chest shows diffuse ground-glass opacities (white arrows) and consolidation (black arrow), within the hilar, perihilar, and lower lobe regions, the lung apices and the costophrenic sulciare spared (white arrows) |
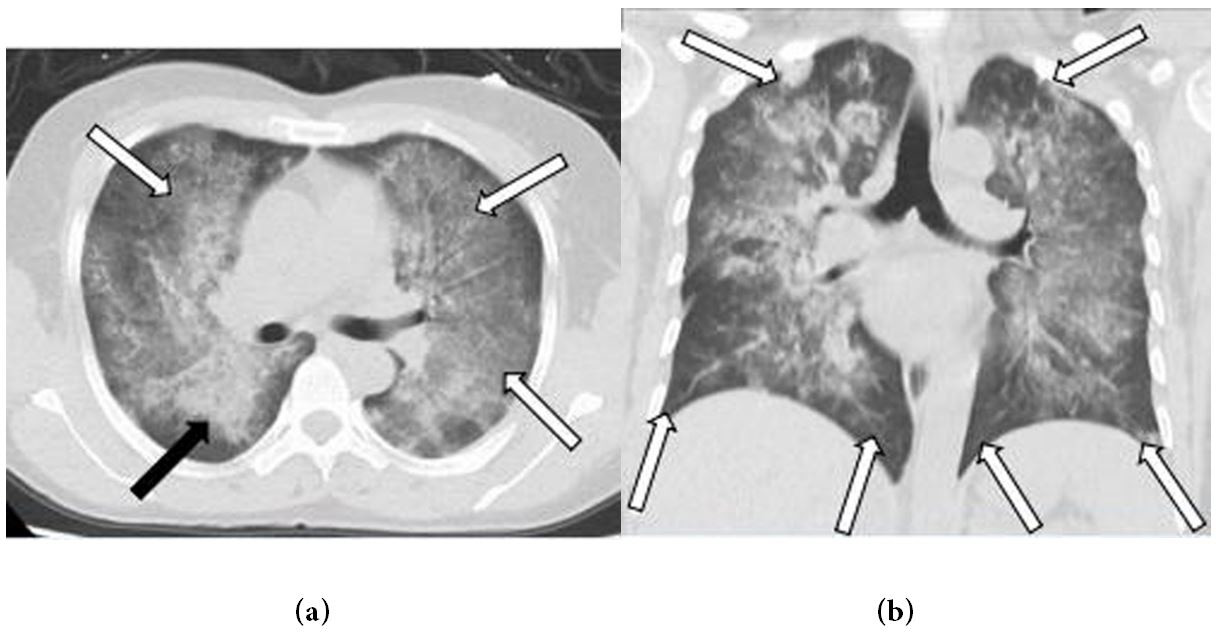 |
| Figure 2: (a) Axial and; (b) coronal unenhanced chest CT after three days treatment with antibiotic demonstrate the lesions are progressive (white and black arrows), especially in bilateral subpleural regions of lung apices |
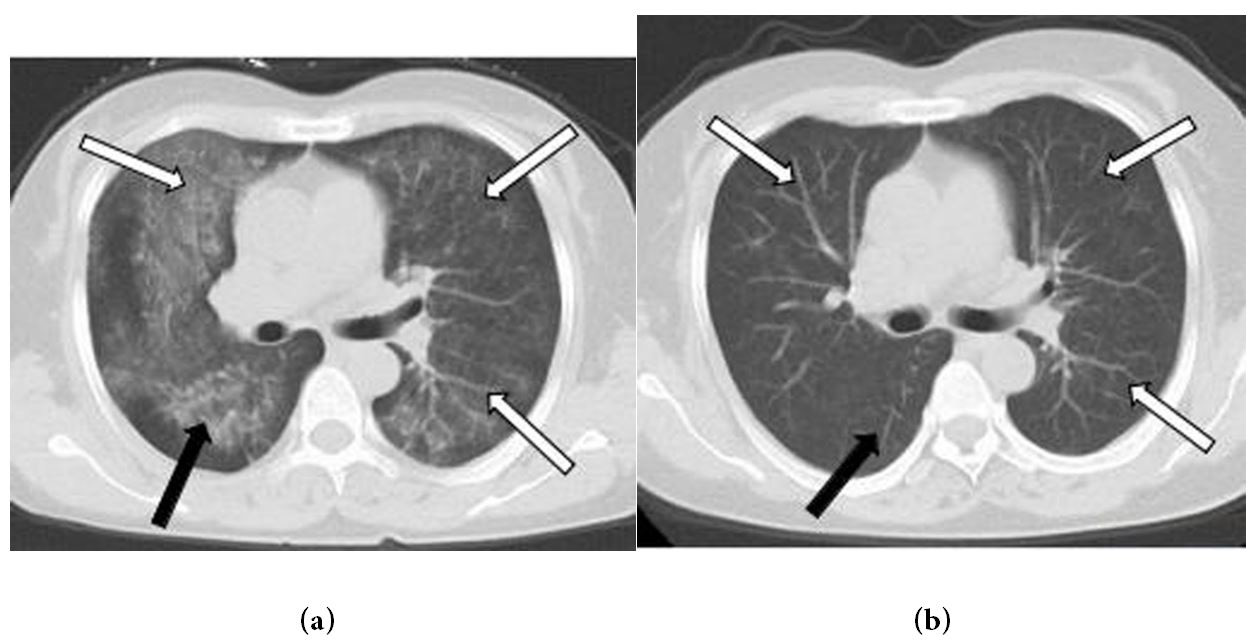 |
| Figure 3: (a) and (b) Axial unenhanced chest CT after one and two weeks’ treatment with hormone and immunosuppressive show the lesions are obviously absorbed (white and black arrows), and vanish (white and black arrows) |




































