Open Access
Case Report
Max Screen >>
ISSN: 2348-9820
Copyright: © 2017 Malik R. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Renal amyloidosis is an important and potentially life threatening complication of pulmonary tuberculosis (PTB) and should be suspected clinically in all patients of PTB presenting with pedal edema, proteinuria, and medical renal disease on ultrasound. Renal amyloidosis may occur in PTB patients at variable intervals. It can occur in patients having disease for relatively shorter duration as well as in chronic cases of PTB with duration of illness often ten or more years. It can occur even if PTB was adequately treated. In tuberculosis, pedal edema may have other causes including malnutrition, anemia and heart failure, which may result in missing amyloidosis in these patients. An increase in awareness of the clinical features of amyloidosis in tuberculosis is important for its early diagnosis and timely assessment. So we report 2 cases of secondary amyloidosis post tuberculosis, one presenting after a long latent period of 25 years and other within few months of diagnosis of PTB.
A 65-year-old smoker male was admitted with complaints of productive cough without hemoptysis and exertional dyspnea for 1 year. He had gradually progressive pedal edema for 6 months and fever for last 20 days. There was no history of skin rash, joint pain, hair loss, or decreased urine output. He had taken treatment for PTB for 6 months 25 years back. He had no history of any other chronic medical illness like diabetes, hypertension, coronary artery disease etc. There was no other history of any other major illness in past or family. On examination he had pitting pedal edema with blood pressure of 110/70 mmHg, temperature of 39 oC, respiratory rate of 20 breaths per minute, and heart rate of 96 beats per minute. Jugular venous pressure (JVP) was not raised. Chest examination revealed bilateral basal crepitations. Other organ system examination revealed no abnormality.
Laboratory investigations revealed the following: hemoglobin, 8.3 gm/L; total white cell count 13,700/μL (differential count: 76% neutrophils, 20% lymphocytes, 2% monocytes, and 2% eosinophils); platelet count 1,89,000/μL; peripheral blood smear was suggestive of normocytic normochromic picture; erythrocyte sedimentation rate was 45 mm/hour, sodium, 134 mmol/L; potassium, 3.8 mEq/L; urea 43 mg/dl; creatinine was 0.8 mg/dL; serum calcium 8.8mg/dl; serum protein 4.4 gm/dL while serum albumin/globulin was 2.4/2 gm/dL. Urine examination showed proteinuria (3+) with 1-2 red blood cells or RBC/high power field or HPF, and pus cells 2-4/HPF, along with hyaline cast. 24-hour urine protein was 5.45 gm/day. Enzyme-linked immunosorbent assays for human immunodeficiency virus, hepatitis B surface antigen, and hepatitis C virus were negative, as also antinuclear antibodies and antibodies to double-stranded DNA negative with normal C3 and C4 levels. Ultrasonography of the abdomen showed kidneys of normal size but raised cortical echogenicity suggesting medical renal disease. Renal biopsy (Figure 1) was done, suggestive of renal amyloidosis (deposition of amorphous material in glomerulus). Congo red staining was also done suggestive of apple green birefringence (Figure 2). Electron microscopy for further characterization of type of amyloid deposit could not be done due to unavailability at our hospital. Electrocardiography was normal. Echocardiogram was normal with normal LV systolic function. High resolution computed tomography of chest showed patchy consolidation with traction bronchiectasis in both lung fields with multiple enlarged lymph nodes in pretracheal, prevascular, and subcarinal and both hilar regions along with hypodense area within nodes suggestive of tuberculosis. Diagnosis was confirmed on sputum examination which was positive for acid fast bacilli (AFB) and subsequently confirmed on AFB culture report as mycobacterium tuberculosis species. The patient was started on category- II antitubercular treatment (ATT, i.e HRZSE x 2 months, followed by HRZE x 1 month and then HRE x 5 months) along with antibiotics for superadded bacterial infection. But during further stay in hospital, the patient deteriorated and finally succumbed to respiratory failure.
An 18-year-old non smoker male was admitted with complaints of fever and productive cough with hemoptysis and exceptional dyspnea for 3 months. After 15 days, he developed gradually progressive pedal edema. There was no history of skin rash, joint pain, hair loss, or decreased urine output. Just 1 month prior to these complaints he had completed anti tubercular treatment (HRZE x 2 months followed by HR x 4 months) for PTB for 6 months with initial diagnosis based on typical pulmonary radiography and sputum for AFB positivity. He had no history of any other chronic medical illness like diabetes, hypertension. There was no other history of any other major illness in past or family. On examination he had pitting pedal oedema with blood pressure of 108/70 mmHg, temperature of 38 oC, respiratory rate of 18 breaths per minute, and heart rate of 90 beats per minute. JVP was not raised. Chest examination-bilateral basal crepitations were present. Other organ system examination revealed no abnormality.
Laboratory investigations revealed the following: hemoglobin, 7.2gm/L; total white cell count 6,500/μL (differential count: 58% neutrophils, 39% lymphocytes, 2% monocytes, and 1% eosinophils); platelet count 2,45,000/μL; peripheral blood smear was suggestive of predominantly normocytic normochromic picture with some microcytes; erythrocyte sedimentation rate was 24 mm/hour. Serum sodium was 138 mmol/L; serum potassium 4.3 mEq/L; blood urea 56 mg/dl; serum creatinine was 1.6 mg/dL; serum calcium 8.1mg/dl; serum protein 4.5 gm/dL while serum albumin/globulin was 2.4/2.1 gm/dL. Urine examination showed proteinuria (3 +) without RBCs or pus cells, along with hyaline cast. 24-hour urine protein was 4.8 gm/day. Chest x-ray was suggestive of infiltrates and cavitary lesion in upper lobes of both lungs. Enzyme-linked immunosorbent assays for human immunodeficiency virus, hepatitis B surface antigen, and hepatitis C virus were negative with antinuclear antibodies and antibodies to double-stranded DNA negative and normal C3 and C4 levels. Ultrasonography of the abdomen showed normal sized kidneys with raised cortical echogenicity. Renal biopsy was done, suggestive of renal amyloidosis including Congo red staining. Sputum culture did not show any organisms. Sputum smear for acid-fast bacillus was negative. CECT chest was suggestive of active tuberculosis with bilateral infiltrates and cavitations in upper lobes with necrotic hilar lymphnodes. Electrocardiography and echocardiogram were normal. Considering the previous history, he was started on category- II ATT (i.e HRZSE x 2 months, followed by HRZE x 1 month and then HRE x 5 months) along with diuretics and ACE Inhibitors (ACEI) to control proteinuria after nephrology consultation. Patient was reviewed 6 weeks later. He was symptomatically better, edema had subsided and even proteinuria decreased to < 500 mg/day. ATT and ACEI were continued. At 6 months follow up, protienuria subsided completely. The patient was lost to further follow up.
Amyloidosis, either primary or secondary, may be defined as a group of heterogenous, chronic infiltrative disorders caused by extracellular deposition of highly organized, insoluble fibrilar proteins in various tissues, that have in common, beta-pleated sheet configuration on X-ray diffraction examination, a fine fibrillar nonbranching appearance on electron microscopy and an apple-green birefringence when examined under polarised light after staining with Congo-red. There are different types of amyloidosis, but a common form worldwide is that which occurs secondary to chronic inflammation, in which amyloid A (AA) fibrils are derived from high circulating concentration of the acute-phase protein serum amyloid A (SAA) [1]. A diverse range of disorders are associated with secondary amyloidosis which includes chronic inflammatory or infectious diseases such as rheumatoid arthritis, osteomyelitis, tuberculosis, chronic bronchiectasis, empyema, ulcerative colitis, carcinomas (most commonly renal cell carcinoma), etc; and most recently in drug abusers, chronic skin infections, transmissible spongiform encephalopathies, Alzheimer’s disease, and type-2 diabetes mellitus [1]. In developed countries rheumatoid arthritis is considered the most frequent underlying inflammatory disease, whereas in developing countries, tuberculosis is still a common underlying cause for renal amyloidosis [1].
The proinflammatory mediators/cytokines like interleukin-1,6, and tumor necrosis factor alpha, which are released in patients with these conditions stimulate the synthesis of SAA in liver and other sites, that subsequently accumulates in renal tissue . But every patient with these chronic inflammatory disorders do not develop AA amyloidosis. This may be due to potential role of other factors such as genetic or environmental influences, specific properties of the precursor protein, macrophage activity, and local tissue factors [1].
Amyloidosis of the kidneys is most common, and is serious because of its ill effects on renal functions. Proteinuria is considered the most consistent finding and may be moderate to severe. The reported incidence of this complication varies from 32-68% [1]. The nephrotic syndrome occurs frequently but not constantly. In some patients, tubular rather than glomerular deposition of amyloid can produce azotemia without significant protienuria. Hypertension is also variable in renal amyloidosis patients with reported incidence of 7-50% [1].
It is often believed that secondary amyloidosis occurs months to years after the onset of the predisposing cause and this interval is variably reported in different studies. It was reported to be between 6 months to 43 years (mean 17 years) by Kennedy, et al., 2 months to 31 years by Mehta, et al., 5 months to 25 years by Shah, et al., 1-30 years (mean 6.92 years) by Chug, et al., 1-6 years (mean 2 years) by Erk, et al., and 2 months to 6 years by Gupta, et al. [1]. A very early onset amyloidosis after the diagnosis of active TB has also been reported by El-Hennawy, et al. [2] and Malhotra, et al. [3], in recent years.
Since the clinical onset of amyloidosis is preceded by a variable preclinical stage, it is reasonable to suspect renal amyloidosis in any patient with known history of PTB presenting with pedal edema, proteinuria and abnormal kidneys on ultrasound.
In both our patients, there was pedal edema and proteinuria in nephrotic range. On ultrasound kidney size was normal in both the patients with increased cortical echogenicity suggestive of medical renal disease. Both the patients had a past history of old treated PTB, and presented with reactivation. Both patients had anemia, which was most likely anemia of chronic disease as peripheral smear was normocytic normochromic. In second patient it was more severe and also associated with microcytes, which could be due to blood loss caused by hemoptysis. Other causes of pedal edema like heart failure were ruled out by normal JVP and echocardiography.
Successful treatment of the underlying inflammatory process can lead to stabilization or improvement in renal function, reduction in protein excretion, and partial resolution of amyloid deposits [4-6]. There have been cases reported in the literature in which there was remission of nephrotic syndrome following treatment of tuberculosis [7,8] but this is certainly rare. Even in our second patient, remission was seen. But most patients with renal amyloidosis ultimately progress to chronic renal failure and once kidney involvement is extensive, anti-TB treatment will not cause any regression in the course of renal amyloidosis. Even despite being treated adequately with effective ATT, patients may present with renal amyloidosis and this could be attributed to posttubercular bronchiectasis or an irreversible process of amyloid deposition that had initiated earlier [9].
Patients with renal amyloidosis who progress to end-stage renal disease can be treated with either dialysis or renal transplantation. Hemodialysis and continuous ambulatory peritoneal dialysis (CAPD) appear to be equally effective, with the limiting factors being the degree of extrarenal amyloid deposition, hypotension with hemodialysis, and peritonitis with CAPD [10]. With careful selection, patients with secondary amyloidosis due to tuberculosis are eligible for renal transplantation with favorable outcome and improved quality of life [9]. Antibiotics and colchicine are effective in preventing and treating infection-related and familial Mediterranean fever-related AA amyloidosis, respectively. Recently, TNF-alpha blockers have emerged as effective agents in inflammatory AA amyloidosis. Eprodisate binds to the glycosaminoglycan binding site on amyloid fibrils, thus targeting amyloid fibril polymerization and tissue deposition. Eprodisate has possible applicability to other types of amyloidosis; the results of a recent randomized trial showed that it may slow the progression of AA amyloidosis-related renal disease but confirmatory studies are necessary [11].
Due to variable preclinical stage, the true interval between the preceding disease and the onset of amyloidosis is not known exactly, so it is reasonable to suspect renal amyloidosis in any patient with pulmonary tuberculosis (past or present infection), presenting with triad of pedal edema, proteinuria and abnormal kidneys on ultrasound, so that early diagnosis is made with timely institution of appropriate treatment. Because even though rare, remission has been reported in renal amyloidosis post ATT as in our second case.

![]()

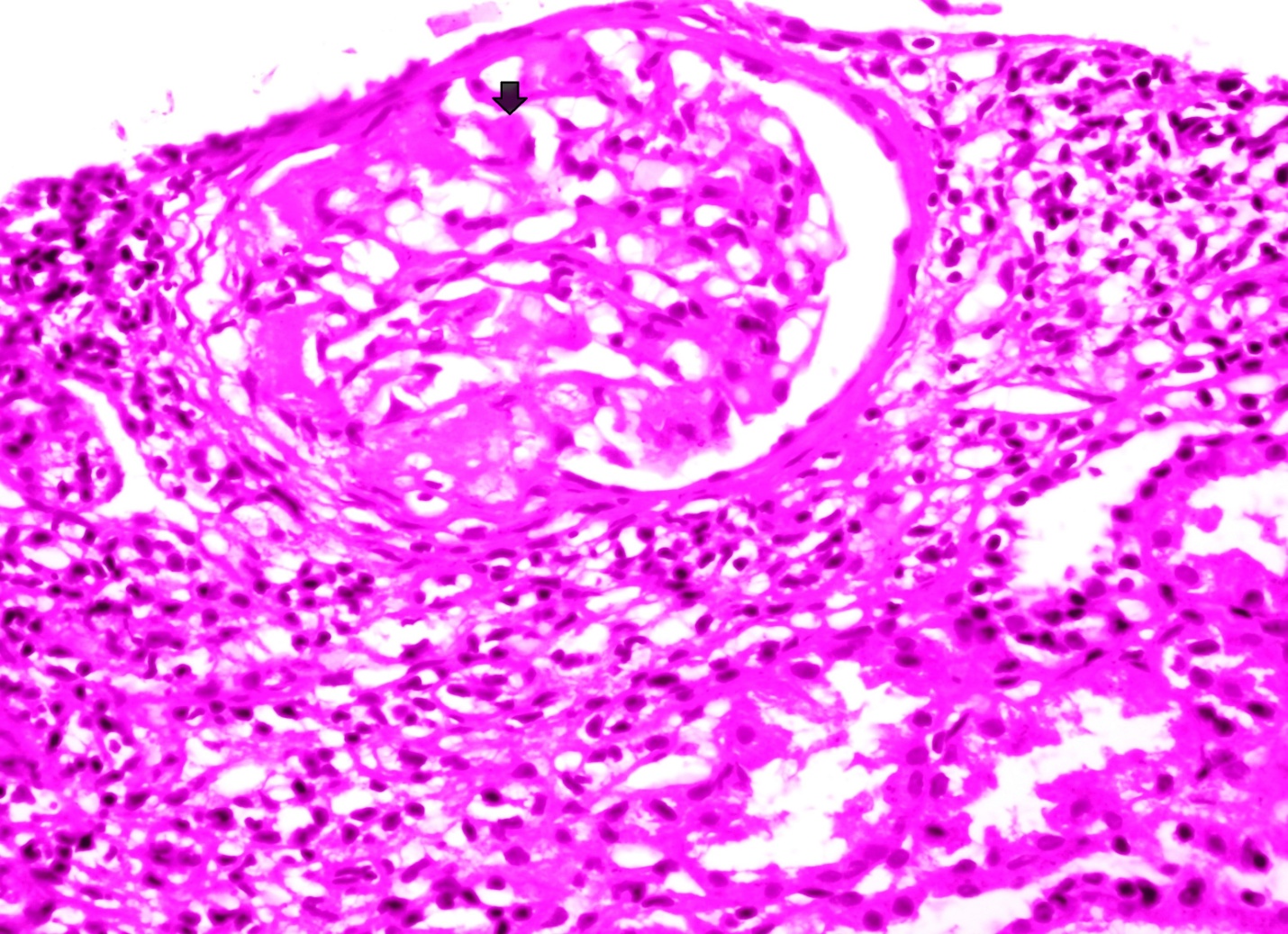 |
| Figure 1: Renal biopsy showing amyloid (pink acellular hyaline material deposit); Amorphous, eosinophilic deposits in the glomeruli: within the mesangial matrix and along the basement membrane of the capillary loops (Arrow) (Hematoxylin and Eosin stain, x 40) |
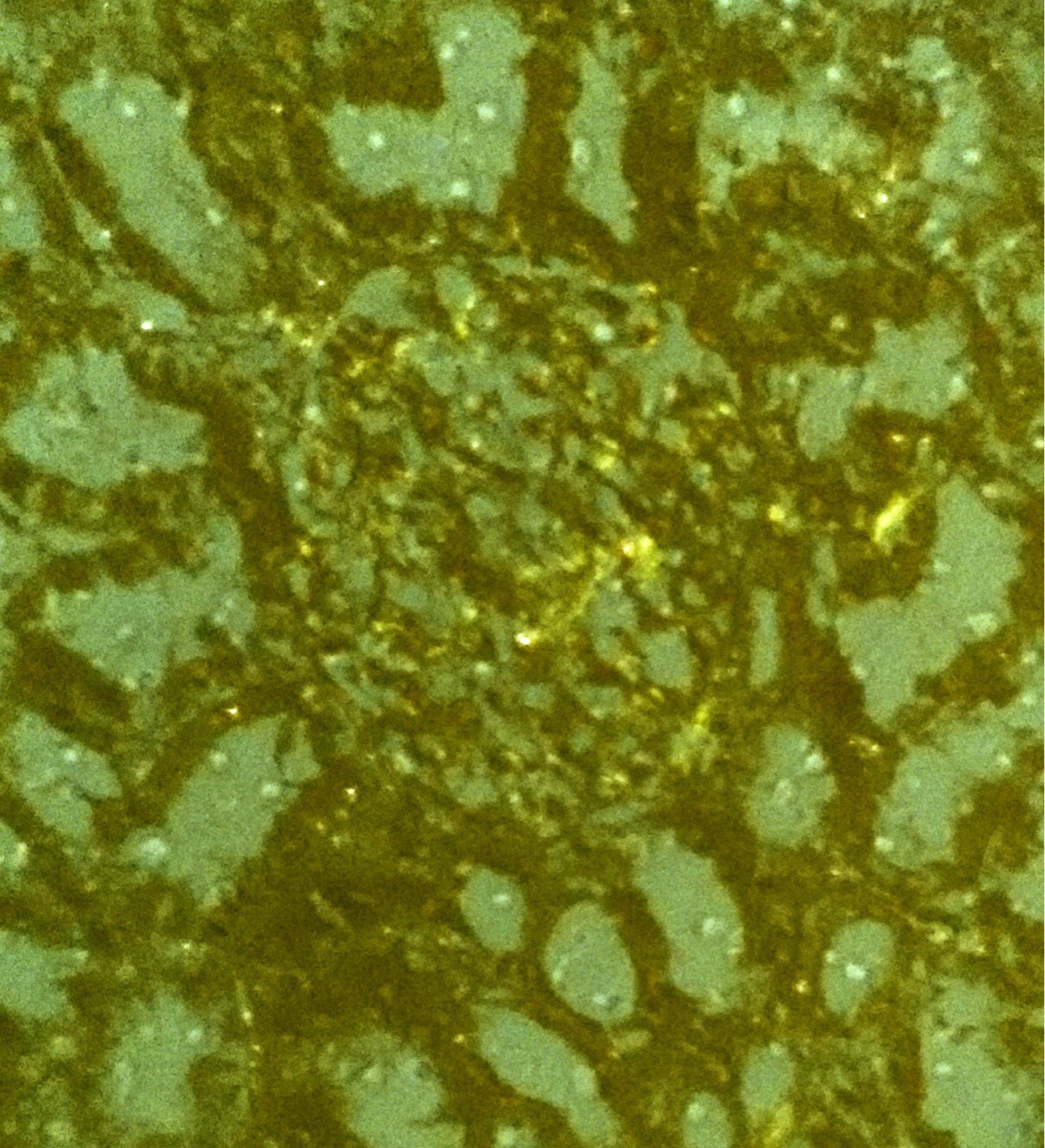 |
| Figure 2: Apple green birefringence of amyloid deposit in glomerulus stained by Congo red (under polarised microscope) |




































