Open Access
Research Article
Max Screen >>
ISSN: 2348-9820
Copyright: © 2015 Bouazzi H. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
X-linked Intellectual Disability (XLID) is an extremely heterogeneous disorder for which many of the causative genes are still unknown. So far, more than one hundred genes of the X chromosome have been found to be altered in males manifesting intellectual disability (ID). NLGN4X is an XLID gene, which has been found, involved in autism and Asperger syndrome involving causative coding mutations. Up to now a few pathological mutations in the promoter and the 5’ UTR have been identified. Here we report a non-syndromic X linked Intellectual disability in two brothers with a novel NLGN4X splicing mutation predicted to have a pathogenic effect by the activation of an exonic cryptic acceptor site, with presence of one or more cryptic branch point(s). This mutation g.1202C>A (Genbank accession number NC_018934.2) was identified through X exome sequencing. It was confirmed by Sanger sequencing. The mother was heterozygous with a skewed X inactivation pattern (100%). She is not affected. This variant was predicted to change the splicing process leading to potential alteration of the mRNA. This mutation segregates with the pathological phenotype in all the affected males. However, it was absent in the non-affected daughter, we suggest that it could be in favor of genotype- phenotype correlation. This study also confirms the efficiency of X exome sequencing for identifying specific genetic conditions not clinically suspected.
Keywords: Autism; Exome sequencing; Intellectual disability; X- inactivation; Mutation
Abbreviations: NLGN4X: Neuroligin; ID: Intellectual Disability; XLID: Chromosome X Linked Intellectual Disability
X-linked intellectual disability (XLID) is a widely heterogeneous group of genetic disorders that involves more than one hundred genes [1]. Neuroligin 4 (NLGN4) is a member of a cell adhesion protein family expressed in postsynaptic neurons that interact with neurexins expressed in presynaptic neurons [2]. Neurologin appears to play a role in the maturation and function of neuronal cells [3]. NLGN4 is an X-liked intellectual disability (ID) gene and it was reported to be altered in Autism spectrum disorder (ASD) which is a condition that affects social interaction, communication, interests and behavior. It includes Asperger syndrome and childhood autism [4]. Mutations in the X-linked NLGN4 gene are a potential cause of (ASD), and mutations have been reported in several patients with autism (OMIM:605309), Asperger syndrome (OMIM:300497), and intellectual disability [5].
Splicing mutations within NLGN4X gene has been confirmed to produce autistic phenotypes [6]. One previous study has reported a lack of one exon within NLGN4X mRNA in a patient belonging to an autistic cohort with a splicing mutation [7]. One more pathological mutation (-335 G>A) was reported to be linked to non syndromic Intellectual disability and ASD [8].
In this study, we report on a novel NLGN4X gene splicing mutation g.1202C>A (Genbank accession number NC_018934.2), between exon 2 and exon 3 respectively (ENSE00001222767/ENSE00002217675- ENSEMBL database) identified through parallel sequencing of all X chromosome exons and predicted by Human Splicing Finder (HSF) to result in activation of an exonic cryptic acceptor site, with presence of one or more cryptic branch point(s) leading to potential alteration of the splicing process. This Splice mutation may lead to potentially abnormal neuroligin in the causation of (ID) and associated signs in our two patients.
The family included two affected brothers with moderate (ID) and one unaffected sister. All family members have been clinically evaluated. Cognitive assessment was achieved in patient II-1 and II-2 using the Wechsler Intelligence Scale Child version four (WISC-IV). Brain magnetic resonance imaging (MRI) was performed in patient II-2. Informed consent for genetic studies was obtained from parents, according to the French bioethics law.
Karyotype and CGH array were performed in both affected brothers.
DNA was extracted from peripheral blood using the standard procedure of phenol chloroform method [9]. Purity and concentration were assessed by NanoDrop ND-1000 Spectrophotometer V3-7 (Thermo Fisher Scientific, Wilmington, DE, USA). DNA from patients II-I and II-II was included in a next generation sequencing project for XLID patients in our institute using SOLiD 5500 sequencer (Life Technologies, Grand Island, NY, USA). Five micrograms of DNA were enriched by micro droplet PCR procedure (Raindance technology, Billerica, MA, USA) to target 11,575 exons.
Sorting and calling of SNP/InDel were performed using SAMTOOL and GATK softwares. Novelty was assessed by filtering the variants against a set of polymorphisms that are available in public databases such as dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 genomes (https://browser.1000genomes.org/in-dex.html), Exome Variant Server (https://evs.gs.washington.edu/EVS/), and ExaC (https://exac.broadinstitute.org/). Only non-synonymous variants or changes affecting splice sites were analyzed. All sequence variants were prioritized by scoring phylogenetic conservation and functional impact (SIFT, Polyphen-2, Splicing Mutation Finder and Mutation taster). Candidate variants were selected and confirmed by Sanger sequencing, using the 3500XL Genetic analyzer and the Big Dye cycle sequencing Kit of Applied Biosystem technology. Forward (5’-GGGTGATGCACGAAATAACG-3’) and reverse (5’-AAGCGGGCAGTTCAGAGATG-3’) primers were designed to amplify the UTR 5’ region of the NLGN4X gene. X-chromosome inactivation study was carried out in patient I-2 (Mother), according to the method of methylation-sensitive PCR and fragment-length analysis of androgen-receptor CAG repeat polymorphism [10].
Patient II-1: This male patient was the child in this family (Figure 1). He has a mild intellectual disability. He was born at term, after a normal pregnancy. Birth length was 51 cm (mean), birth weight was 3.5 kg (+0.5SD), and birth head circumference was 35 cm (mean). Apgar score was 10 over 10 minutes. His early psychomotor development was delayed. Started walking at 18 months. At the age of 9 he has a dyspraxia with learning difficulties associated with visual hallucinations. He had great difficulties of morization memorization. He did not know his date of birth or the surname of his grandparents nor the time situation; however, he could do his daily tasks alone. He did not experience any seizure episode. An EEG was performed at 7 years old and was normal. Brain MRI, performed at the age of 7 years, and did not reveal any alteration. He had no facial dysmorphic features. His intellectual quotient (IQ) was 50. He has been treated with Ritalin for dyspraxia and being followed in a French speech and behavior rehabilitation center. CGH-array and karyotype studies were normal.
Patient II-2: This 10-years –old male (index case) was the second of a sibling of three (Figure 1), born to unrelated young parents. He was born at term after a normal pregnancy. Birth weight was 3.6kg (+0.7SD), birth length was 52cm (mean), and birth head circumference was 35.5cm (mean). Apgar score was 10 over 10 minutes. There have been no difficulties in the neonatal period and psychomotor acquisitions were done in time without any language delay. His first neuro-cognitive difficulties appeared during schooling. At eight years old, he did not acquire reading, nor writing, hehad severe dyslexia associated with impairment of semantic processing. He had great difficulties of memorization (He did not know his date of birth or the surname of his grandparents nor the time situation). He was easily distressed, mainly in situations of failure. He had an attention deficit-hyperactivity disorder and panic and rage attacks involving agitation and impulsiveness associated with failure in concentration. He presented a dyscalculia and a spatial visual disorder without any signs of dyspraxia. Otherwise his neurological exam was normal and his intellectual quotient (IQ) was assessed for 55. Besides that he has no behavior disorder and no insomnia. His feeding was normal. He had no significant dysmorphic features (Figure 2AB). At 10- years- old, his weight was 33 Kg (mean), height 136 Cm (mean). MRI was performed at 8- years- old and did not reveal any alteration. Metabolic and endocrine analysis was normal. Karyotype and array-CGH did not show any pathogenic chromosomal imbalance.
Sequencing of X-exome from patient II-1 and patient II-2 identified one splicing variant in NLGN4X gene that could be considered as potentially pathogenic according to our filters. There were no more other predicted pathological mutations of X-linked genes identified in two affected brothers.
The variant was a g.1202C>A substitution within the splicing site of intron 2 between exon 2 and exon 3 of LGN4X gene (GenBank accession number NC_018934.2) leading to the substitution of a Cytosine to an Adenine (IVS2+8C>A). This mutation was absent from public databases of control individuals (Exome Variant Server, 1000genomes, dbSNP135, and ExaC) and in > in-house 200 X-exomes of index patients from other X-LID families. This mutation was confirmed by Sanger sequencing in the two affected boys as well as in the mother, but it was not studied in the unaffected sister. This substitution was predicted by Human Splicing Finder software (HSF) to result in the activation of an exonic cryptic acceptor site, with presence of one or more cryptic branch point(s) leading to potential alteration of the splicing process. Predictions with Mutation Taster were in favor of a change of a splice site. X-chromosome inactivation pattern was completely skewed in the mother (100%) who was heterozygous and asymptomatic.
X-exome sequencing applied to the X-LID family reported here brought out novel variant occurs in the late exonic positions in NLGN4X gene (NC_018934.2:g.1202C>A)-Genbank/ (IVS2+8C>A)- ENSEMBL. This splice site mutation could be considered as potentially pathogenic. It was confirmed by Sanger sequencing. It segregated with the pathological phenotype of the two affected boys.
NLGN4X gene encodes a protein which belongs to a family of neuronal cell surface proteins [11]. It acts as splice site-specific ligands for beta-neurexins and may be involved in the formation and remodeling of central nervous system synapses [12]. Mutations in this gene have been associated with autism (OMIM300495), Asperger syndrome (OMIM300497) and Tourelle syndrome (OMIM 137580) [3].
So far several pathological mutations have been reported within NLGN4X gene and have been associated with (ID) and a wide clinical variability [13,14].
The alteration of NLGN4X gene in Autism is characterized by impairments in reciprocal social interaction and communication, restricted and stereotyped patterns of interests and activities, and the presence of developmental abnormalities by 3 years of age. Most individuals with autism also manifest moderate to mild intellectual disability [15].
NLGN4X was reported to be altered in Asperger syndrome which involves features similar to autism. Affected individuals exhibit qualitative impairment in social interaction, as manifest by impairment in the use of non-verbal behaviors such as eye-to-eye gaze, facial expression, body postures and gestures, failure to develop appropriate peer relationships, and lack of social sharing or reciprocity. Patients also exhibit restricted, repetitive and stereotyped patterns of behavior, interests and activities, including abnormal preoccupation with certain activities and inflexible adherence to routines or rituals. Despite this similarity, Asperger syndrome is distinguished from autism by the higher cognitive abilities and a more normal and timely development of language and communicative phrases [16].
Previous studies have reported that pathological mutations within this gene were identified and found associated with Tourette syndrome involving neurobehavioral disorder characterized particularly by motor and vocal tics and associated with behavioral abnormalities. Tics are sudden, brief, intermittent, involuntary or semi-voluntary movements (motor tics) or sounds (phonic or vocal tics) [17].
Despite the heterogeneity of clinical features associated with the alteration of NLGN4X gene in these different syndromes, most patients shared a common core of clinical features represented by moderate to mild (ID) and behavior disorder. However neither social interaction impairment which is a common criterion for Autism nor stereotypy that was commonly delineated in Asperger syndrome was noticed in our present family. Likewise motor and vocal tics which are constant in Tourette syndrome were not detected in this study. Nevertheless, patient II-2 of this family had attention deficit-hyperactivity disorder and panic and rage attacks. These signs were reported in Asperger syndrome. The mild intellectual disability of our patients was shared with most patients of previous studies that have reported causative NLGN4X gene mutations.
Alterations in NLGN4X gene were confirmed to cause a wide heterogeneous pathological features [18]. Patients of this study inherited a novel NLGN4X gene splicing mutation (g.1202C>A) from an asymptomatic mother. The X-inactivation pattern of the mother was completely skewed (100%). The mutation segregates with the pathological phenotype and was predicted to result in activation of an exonic cryptic acceptor site (ACGGTAGCTCAG) with presence of one or more cryptic branch point(s) (Figure 3) leading to potential alteration of the splicing process. The proximity of this mutation from exon 2 and its location in the splicing donor site (IVS2+8C>A) may lead to a failure in the normal splicing process and then to an abnormal NLGN4X transcript, thus an expression of a pathological phenotype.
In the normal splicing process, exon2 and exon3 are situated in the 5’UTR (untranslated region) within a regulatory promoter sequence. Therefore, we thought that the new mutation could lead to a failure of this process and we suggest that this variant could be the cause of the pathological phenotype. However to confirm this suggestion, additional function studies should be performed. The assessment of mRNA quality could help to confirm the effect of this mutation. Expanded sequencing for more related members of this family would give more details about the inheritance of the pathology. The study of X inactivation pattern in the sister II-3 (Figure 1) would be helpful to strengthen our suggestions.
The involvement of the NLGN4X gene in intellectual disability has been confirmed. There are several pathological mutations known in this gene. Our substitution splicing mutation (g.1202C>A) segregates with the pathological phenotype in all affected patients and predictions were in favor of pathological effect. Although functional assessments of NLGN4X gene were not performed in this study, this result could bring further insight about the phenotypic spectrum of NLGN4X mutations and suggests that mutations in different regions of the gene might cause variable phenotypes. However, more additional studies are required to confirm the genotype-phenotype correlation.
We are grateful to the patients. We thank the team of the Necher University Hospital High-throughput sequencing platform for the technical support in performing X-exome sequencing. This work was supported by the Imagine Foundation (Necker Hospital) and by INSERM U781. We especially appreciated the clinical criticism and advices of Doctor Karim Bengaoua from the ESTREE private hospital.

![]()

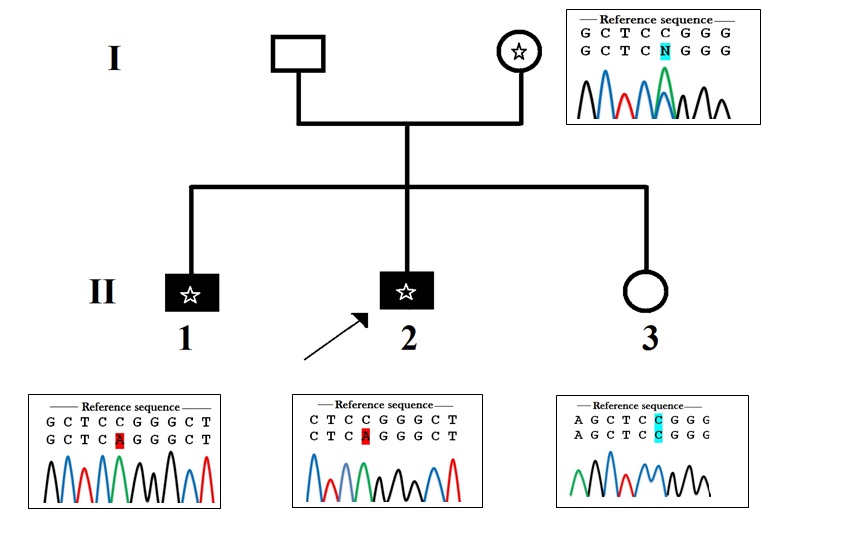 |
| Figure 1: Pedigree of the family. Black symbols indicate intellectual disability. Asterix indicates the presence of the mutation g.1202>A. The arrow shows the index case. A circle with an Asterix represents the carrier female. Sanger sequencing electrophoresis profile shows that the daughter hasn't inherited the mutation; she is homozygous (C/C). The mother is heterozygous (C/A) and both affected boys have inherited the muted allele (A) from their mother |
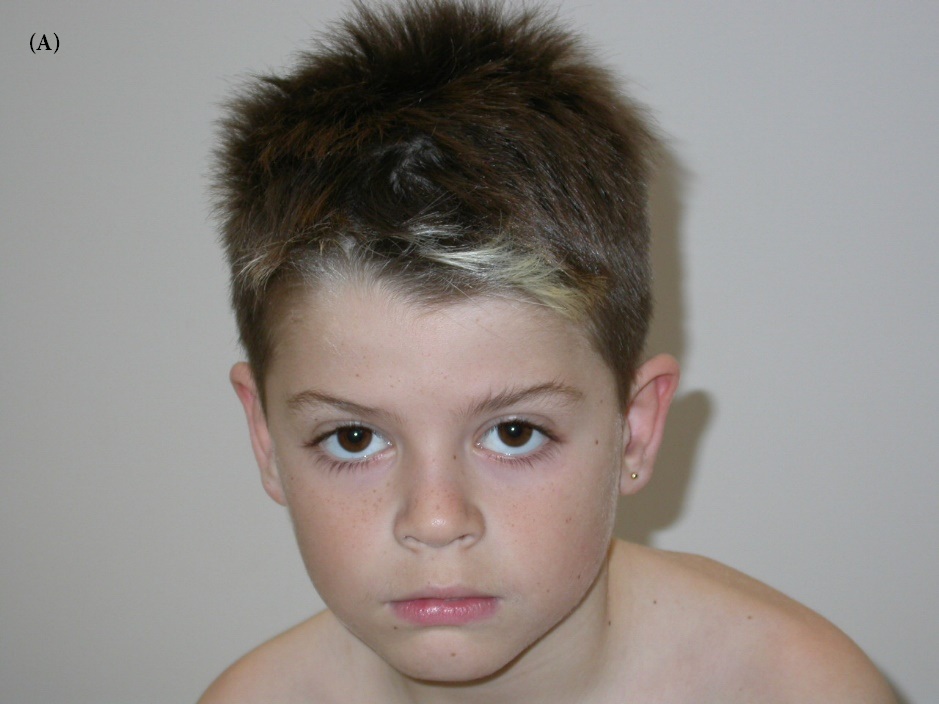 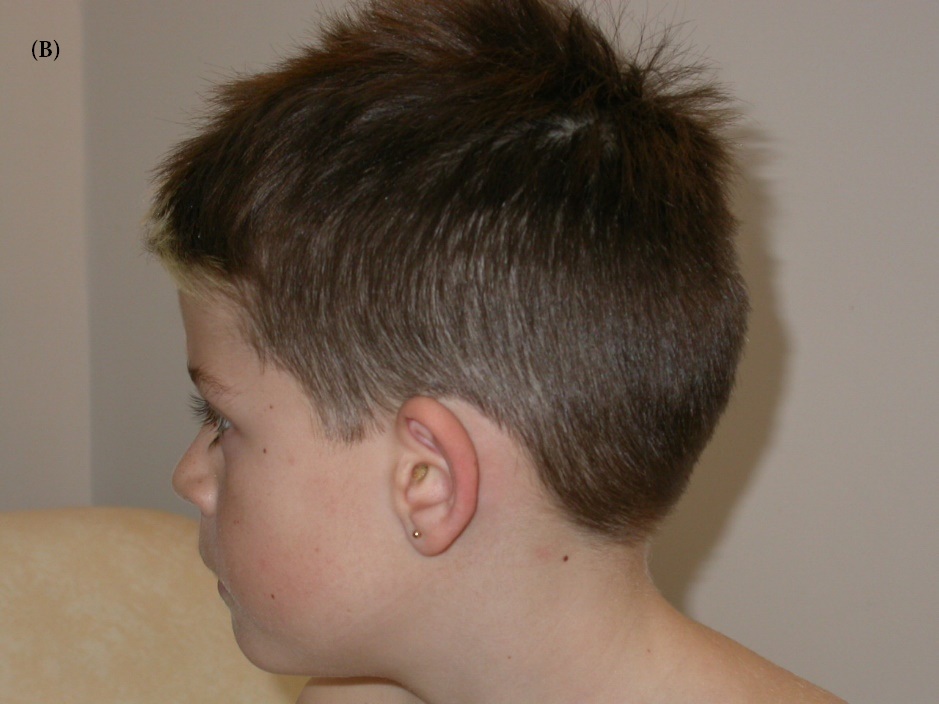 |
| Figure 2AB: Photographs of patient II-2, the index case.2A, 2B respectively profile and face photos, the patient has a mild intellectual disability associated with attention deficit-hyperactivity disorder and a failure in memoriza tion. He has no significant dysmorphism |
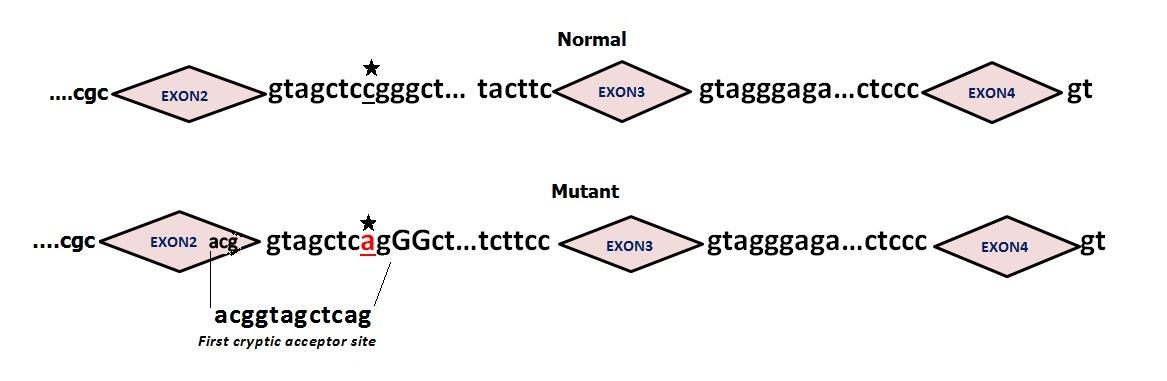 |
| Figure 3: The novel mutation NC_018934.2 g. 1202C>A (IVS2+8C>A) indicated by an asterisk results in a new splice site (ACGGTAGCTCAG) between exon2 and exon3 of the NLGN4X gene (Ensembl sequence reference: ENSG00000146938). This mutation occurs in the promoter region and it is predicted to be pathogenic by altering the normal splicing process |




































