Open Access
Case Study
Max Screen >>
ISSN: 2348-9820
Copyright: © 2014 Singh P. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
Adrenocortical carcinoma is a rare endocrine tumour with poor prognosis. Herein we report a case of adrenocortical carcinoma which was non-functioning, and non-metastasizing. A female patient presented with vague abdominal pain and palpable lump on examination. A large sized adrenal mass on Ultrasonography (USG) and computed tomography was revealed. USG guided fine needle aspiration cytology was performed, the smears were highly suspicious of malignancy. Tumour was surgically excised and histopathology confirmed the final diagnosis of stage II adrenocortical carcinoma. No metastasis was seen. Rarity of non-functioning adrenocortical carcinoma prompted us to report this case.
Keywords: Adrenocortical tumour; Adrenocortical carcinoma; Non-functioning
Adrenocortical carcinoma (ACC) is a rare neoplasm with poor prognosis and with an incidence of one in one million populations. There is a slight female predilection (58.6%) [1]. It has a bimodal age distribution with peaks in the first and fifth decades of life. About 60% of patients present with symptoms of excessive hormone production. However, ACC is usually non-functional when it occurs in adults. Most patients with non- functional ACC present with stage IV disease with large abdominal masses [2,3]. Clinically silent or biochemically and clinically asymptomatic adrenal masses, also known as incidentaloma, are incidentally discovered lesions, when non-invasive imaging methods such as Ultrasonography (USG), Computed Tomography (CT), Magnetic Resonance Imaging (MRI) scan are performed for reasons other than known or suspected adrenal diseases [4]. Most ACCs are large and weigh from 0.1 to 1kg [5]. The prevalence of tumours with a diameter > 1.5 cm is 1.8% and with diameter > 6 cm is 0.025% [6]. It represents 0.02% of all cancers and causes 0.2% of all cancer deaths [7].
Herein, we present a case of non-functioning adrenocortical carcinoma in a 45 years old female.
A 45 years old female patient presented with complaint of dull, vague abdominal pain. It was not associated with any other complaint. On clinical examination, a lump was palpable in left hypochondrium with mild tenderness. There was no pallor, icterus and cyanosis. Pulse rate and blood pressure were normal. Urine biochemistry and microscopic examination were normal. Serum electrolytes and alkaline phosphatase value were within normal limits. Levels of adrenal hormones (urinary vanil mandelic acid and serum aldosterone) were within normal limits.
USG abdomen revealed a heterogeneous solid mass in left suprarenal region. No other abnormality was detected.
CT scan revealed a large heterogeneous enhancing mass at the superior border of left kidney (Figure 1a). It was indenting the superior pole of left kidney. Both kidneys were normal in size.
USG guided fine needle aspiration cytology (FNAC) was performed, which revealed atypical cells arranged in sheets, loosely cohesive clusters, groups as well as dispersed singly. The nuclei of these cells were round to oval to spindle shaped revealing moderate pleomorphism, coarsely clumped irregular chromatin and 1-2 prominent nucleoli at places. Cytoplasm was moderate in amount, fragile and well-defined. Background revealed scant necrosis.
In view of above findings, the tumour was reported to be malignancy of adrenocortical origin.
Exploratory laparotomy was done and complete surgical excision was carried out. The mass could be separated from left kidney and other surrounding tissues clearly.
On gross examination, the tumour mass weighed 725 gms and measured 13×9×8 cms. It was well encapsulated. Cut surface revealed a variegated appearance (Figure 1b).
Histopathological examination revealed adrenocortical carcinoma with areas of necrosis, haemorrhage, high mitotic rate and atypical mitoses (Figure 2 a,b). Capsular invasion was seen. Pericapsular tissue was free from tumour.
Histopathological diagnosis of adrenocortical carcinoma was made.
Adrenocortical carcinoma is a rare, aggressive and heterogeneous tumour with poor prognosis [8]. It represents 0.02% of all cancers and causes 0.2% of all cancer deaths [7]. Tumours arising from adrenal cortex are rare, ACCs are most common among them and account for 6% of adrenal tumours [9]. It has a bimodal age distribution with peaks in the first and fifth decades of life [2,3].
ACC is classified as functional and non-functional based on the hormone syndromes they produce. Functional tumours are common and are detected earlier than non-functioning tumours due to production of hormones and associated clinical signs as well as symptoms. Non-functioning tumours remain undiagnosed till late and mostly present with a large mass and metastatic disease due to their silent nature [10,11].
Non-functional ACCs are less common. ACCs are slightly more common in females than males. Non -functional ACCs occur in older age group (>30 years) and are more common in men (3:2 male to female ratio), while functioning tumours are more common in women (7:3 female to male ratio) [12]. However, other studies showed that non-functional ACCs are slightly more common in women (58.6%) than they are in men (41.1%) [2,3].
Data in literature corroborates with the present case. As in our case the tumour was non-functional and presented at the age of 45 years. The only presenting symptom was dull vague pain in left hypochondrium and the only clinical finding was lump palpable in left hypochondrium. All the other investigation like liver function test, kidney function test, serum adrenal hormones levels were within normal limits.
Primary adrenocortical carcinomas are large tumours usually measuring more than 5 cms at presentation. Larger the tumour more is the chance of it being malignant. Because they are large, the site of origin is often difficult to determine. CT scan plays an important role in characterising the organ of origin and in defining the extent of the primary as well as assessing the presence of metastatic disease [9]. Mass lesions found incidentally by upper abdominal CT examination, so called incidentaloma may be primary adrenal tumours, metastatic malignancies or non-neoplastic lesions with an equal probability of the neoplasm being metastatic or primary. The efficacy of image –guided FNB in the diagnosis of adrenal lesions has been demonstrated in several studies [13].
The size of the tumour in the present case was >5 cms on USG, which was suspicious of malignancy. USG guided FNAC smears in this case revealed high cellularity, necrosis, nuclear pleomorphism and prominent nucleoli, which were indicative of malignancy.
Keeping in view the above findings, the adrenal mass was removed, with clearance of margins.
Differentiation between benign and malignant adrenal lesions is based on macroscopic features (tumour weight, haemorrhage, and breached or intact tumour capsule) and a microscopic diagnosis using the Weiss score, the most commonly used. The score is determined by adding up the following features: mitotic rate, atypical mitosis, nuclear grade, low percentage of clear cells, necrosis, diffuse tumour architecture, capsular invasion, sinusoidal invasion and venous invasion. A score higher than 3 is suggestive of malignancy [14].
In our case nuclear atypia, frequent and atypical mitoses, capsular invasion and necrosis was evident suggesting a diagnosis of adrenocortical carcinoma (Figure 2 a,b).
Non-functioning adrenal tumours remain a diagnostic challenge for early diagnosis and successful management, as there are no early signs and symptoms. In majority of the cases, the tumour has either invaded adjacent organs or already metastasized to distant organs at the time of initial diagnosis [9]. In this case, the distant metastasis was not observed, only capsular invasion was present. Pericapsular tissue was free of tumour.
In 2008, Fassnacht et al suggested modifications included in the European Network for the Study of Adrenal Tumours Classification. This updated staging system groups ACC as follows: T1: tumours <5cm, T2: tumors >5 cm, T3: tumour infiltration of surrounding tissues, T4: tumour invasion of adjacent organs, vena cava or renal vein, N0: no nodes, N1: positive lymph nodes, M0: no metastasis, M1: distant metastasis. Therefore stage 1 includes T1 N0 M0; stage 2 is T2 N0 M0; stage 3 is T1-2, N1, M0 or T3-4, N0-1, M0; and stage 4 is any T, any N, and anyM1 [15]. In the case under discussion, the tumour was in stage II (T2N0M0).
The only curative therapy is complete surgical excision when tumours are small and localized. Complete surgical resection allows the best prognosis; an en bloc resection of the tumour and surrounding structures if indicated is recommended [3,14,16]. If local invasion or metastasis has not occurred, it is difficult to define malignancy on histological examination. Predictors of poor survival after resection include lesions larger than 12 cms, 6 or more mitotic figures per 10 HPFs on microscopy and tumour hemorrhage [16].
The tumour in this case was in stage 2. Complete surgical excision including surrounding tissues was done. Patient is under follow-up and no recurrence of tumour has been reported till date, despite presence of all the predictors of poor survival.
Other features suggestive of ACC are IGF-II over expression, allelic loss of chromosome 17p13 and high level of CyclinE [14].
I thank the on call junior resident Dr Sundar Rajan and the theatre staff whose immense contribution was noteworthy.

![]()

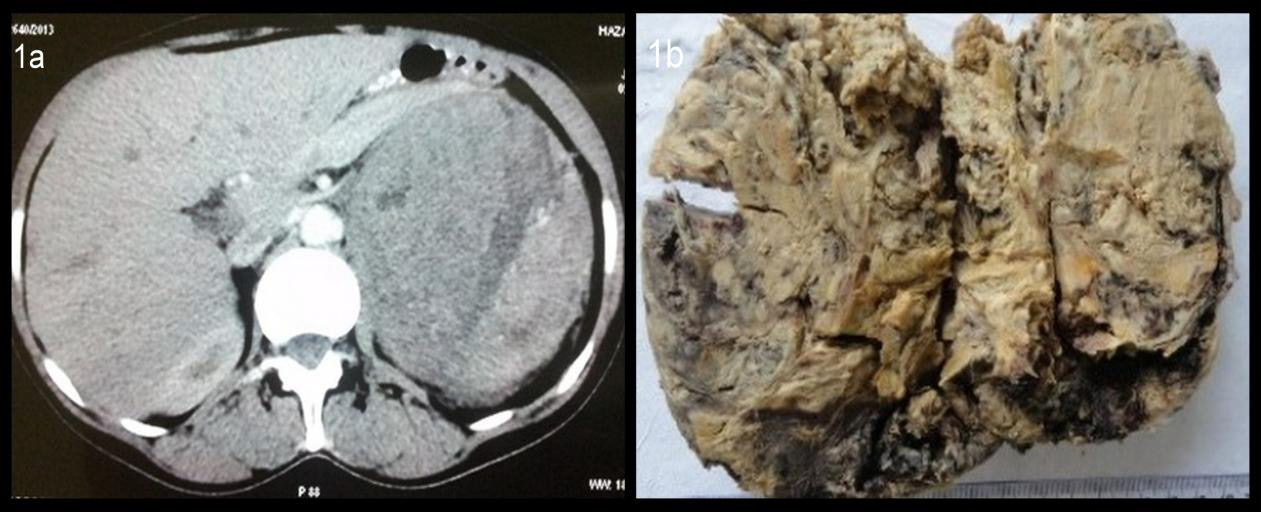 |
| Figure 1a: Computed tomography showing adrenal mass Figure 1b: On Gross examination a grey white soft tissue mass was identified measuring 13×9×8 cm |
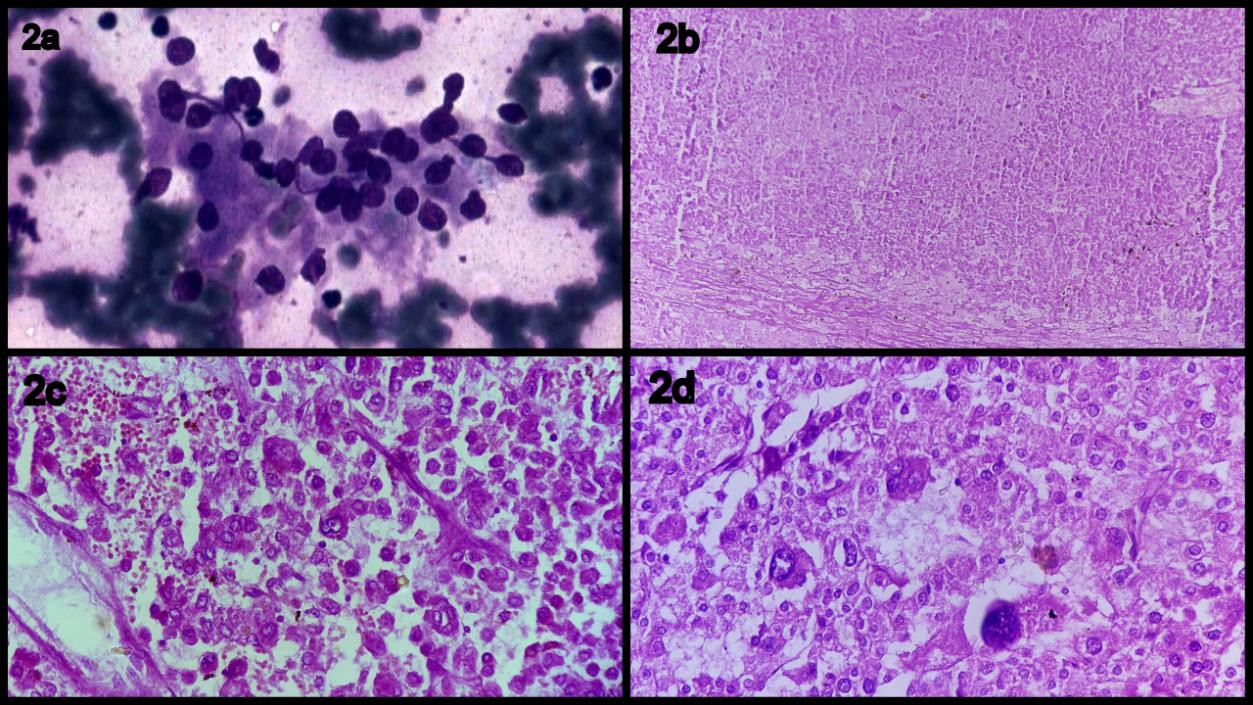 |
| Figure 2a: Fine-needle aspiration features of adrenal cortical carcinoma showing bland tumor cells with minimal nuclear pleomorphism that are plasmacytoid with eccentrically located nuclei, reminiscent of a low - gradeneuroendocrine tumor. Figure 2(b-d): Photomicrograph showing histomorphological features of adrenocortical carcinoma (b: low power), (c: hemorrhage & necrosis) (d: mitosis) |




































