Open Access
Research Article
Max Screen >>
ISSN: 2575-551X
Copyright: © 2016 Rosete-Reyes A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
In Mexico, the immediate-release formulation of omeprazole 20 mg in combination with 1100 mg of sodium bicarbonate (oral capsules) is indicated for patients over 18 years for the treatment of gastritis, reflux, heartburn and the sensation of emptiness caused by the excessive acid production.
The bioequivalence of a test formulation was evaluated with respect to the corresponding reference drug formulation, which was administered as a capsule.
This study had a randomized-sequence, single-dose, single-blind, two-period crossover design and was performed under fasting conditions with 32 healthy Mexican adult subjects, including both genders. There was a seven-day washout period.
The study formulations were administered after an overnight fast (10-hours) , and blood samples were collected at baseline and 0.33, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 6, 8 and 10 hours after administration. The omeprazole concentration in plasma was determined using HPLC coupled with mass spectrometry (MS/MS).
The test and reference formulations were considered bioequivalent if the 90% CIs for the geometric mean test/reference ratios were within a predetermined range of 80% to 125%. The 90% CIs for omeprazole Cmax, AUC0–t and AUC0–∞ were 84.08% to 104.47%, 87.42% to 108.18%, and 87.94% to 108.15%, respectively. These results satisfied the regulatory requirements for assuming bioequivalence, based on the rate and extent of absorption.
Keywords: Omeprazole; Bicarbonate; Combination; Bioavailability; Bioequivalence
Omeprazole is a proton pump inhibitor that is used to treat various acid related diseases [1,2]. Omeprazole has the disadvantage of being acid labile, hence, this drug is easily inactivated in the acidic environment of the stomach [3].
Most of the current oral solid dosage forms of omeprazole require enteric coating to protect the active ingredient against acid degradation. However, the enteric coating delays the absorption of omeprazole and the initial antisecretory effect. The lack of a rapid therapeutic response may contribute to patient dissatisfaction with treatment and may lead to unnecessary increases in dose or inappropriate switching to alternate drugs [4].
This issue has led to the development of immediate-release formulations based on the combination of omeprazole with sodium bicarbonate [5]. The addition of the antacid sodium bicarbonate has been shown to effectively maintain the stability of the omeprazole in gastric fluid because it raises the gastric pH rapidly, thus protecting the omeprazole from gastric acid degradation [6].
In addition, these immediate-release formulations have been proved to provide a more rapid absorption of omeprazole with shorter times to peak plasma concentrations than do enteric-coated formulations [5].
In Mexico, the immediate-release formulation of omeprazole 20 mg in combination with 1100 mg of sodium bicarbonate (oral capsules) is indicated for patients over 18 years for the treatment of gastritis, reflux, heartburn and the sensation of emptiness caused by excessive production of acid [7].
The sponsor of this study (Laboratorios Liomont, S.A. de C.V.) was interested in obtaining the marketing authorization for omeprazole 20 mg in combination with 1100 mg of sodium bicarbonate as a capsule formulation (test formulation) in Mexico (Inhibitron Twit®, Laboratorios Liomont, S.A. de C.V., Mexico City, Mexico).
Therefore, this study aimed to investigate the bioavailability and the bioequivalence of a test formulation containing 20 mg of omeprazole in combination with 1100 mg of sodium bicarbonate in comparison to the corresponding reference drug formulation (Trinsica® 20 capsules, GlaxoSmithKline Mexico, S.A. de C.V.).
A literature survey using PubMed, MEDLINE and Google Scholar data through May of 2016 revealed no hits regarding the combination of the following terms: omeprazole, sodium bicarbonate, bioequivalence, bioavailability, pharmacokinetics, capsules, Mexico, Mexican and population.
The study protocol (P434S034V003) and the informed-consent documents were reviewed by an independent ethics and research committee (Comité de Ética e Investigación para Estudios en Humanos, Mexico City, Mexico). This study was approved on February 8, 2013. We also received approval from the Federal Commission for Protection against Sanitary Risks (Comision Federal para la Proteccion contra Riesgos Sanitarios [COFEPRIS]) on April 17, 2013.
The study was conducted according to the Declaration of Helsinki (and its amendments) and the International Conference on Harmonisation for Good Clinical Practice Guideline.
The subjects were informed about the procedures in the study, the duration of the study, and the anticipated risks and potential discomfort associated with the study by the principal investigator. All subjects gave written informed consent. The clinical stage of the study was performed from June 10 to June 18 of 2013 and the analytical stage from September 2 to September 11 of 2013.
Healthy Mexican adults of both genders were who were aged 18 to 55 years were considered eligible for this study. The subjects were recruited from the volunteer database at the Center of Pharmacological and Biotechnology Research (clinical unit) in Medica Sur Hospital, Mexico City, Mexico.
The health conditions of all of the candidates were evaluated. The evaluation included an interview and a physical examination (blood pressure [BP], heart rate, weight, height, temperature and respiratory rate). The diagnostic testing that included a 12-lead electrocardiogram and chest radiography. The laboratory tests included hematology and blood chemistry, urinalysis, and tests for alcohol, drug-abuse and a pregnancy test in women. The employed serological tests included hepatitis B and C, as well as HIV antibodies. All tests were performed at Medica Sur Hospital with certification from the Mexican government and the College of American Pathologists.
Systolic and diastolic BP were determined with a sphygmomanometer (Tycos; Welch Allyn, Skaneateles Falls, NY). The BP cuff was applied to the right arm and the reading was taken with the subject in a seated position. Subjects were excluded if their laboratory values were considerably outside of the reference range and/or if all tests were not completed.
Prior to the enrollment of the subjects, the laboratory data were reviewed and approved by the clinicians.
A single-dose randomized-sequence, single-blind, two-period crossover design under fasting conditions was used. The subjects were admitted to the clinical site on the day before drug administration, and were randomly assigned to one of the following two sequences in a 1:1 ratio: the test formulation capsules containing omeprazole 20 mg in combination with 1100 mg of sodium bicarbonate (lot 237D0006; expiration date March 2015) followed by the reference formulation capsules containing omeprazole 20 mg in combination with 1100 mg of sodium bicarbonate (lot 1204700034; expiration date December 2013), or vice versa. The assignments were made by a pharmacist using a computer-generated table of random numbers in the presence of quality assurance personnel at the clinical unit.
To ensure reliable baseline plasma measurements, participants underwent a 10-hour overnight fast. Based on the reported plasma half-life of omeprazole, which ranges from 0.6 to 1 hour, a seven-day washout period was considered to be appropriate because it exceeds the seven half-lives required by COFEPRIS, as well as the number of half-lives required by the FDA and the EMA [8-10].
Blood samples were drawn for baseline plasma determinations in the following way. An 18-GA x 1.16 in (1.3 x 30 mm) indwelling angiocatheter (BD-InSyte, Becton Dickinson Ind. Cir. Ltda, Minas Gerais, Brazil) was inserted in a suitable forearm vein and a 7.5-mL blood sample was drawn into a lithium heparin-treated vacuum tube (S-Monovette, Sarstedt AG & Co., Nümbrecht, Germany).
The subjects were administered a single capsule of the test or the reference formulation with 250 mL of water. Additional blood samples were drawn at 0.33, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 6, 8, and 10 hours after administration.
During hospitalization, the subjects were kept under medical surveillance, and during the washout period participants maintained contact with the clinicians to report any adverse events (AEs).
Plasma was obtained by centrifugation (3000 rpm for 15 minutes at room temperature) and stored at -70±10 °C (until the samples were transported to the analytical unit where they were stored at -75±5 °C until the time of analysis). After a seven-day washout period, the subjects returned to the clinical unit, where the alternative formulation was administered as described for the first treatment period.
The subjects were asked to refrain from water and food intake for three hours after the study drug administration. For each treatment period, the subjects diet consisted of two standardized meals (2497.1 kcalorie/day), which were ingested at 3.25 and 8.25 hours after the study drug administration.
Omeprazole (lot: JOH412) and pantoprazole sodium (internal standard, lot: H1M239) reference standards were obtained from the USP (Rockville, MD). All solvents (including water) were HPLC-mass spectrometric grade (Avantor Performance Materials, Inc., Phillipsburg, NJ and Honeywell International Inc., Morristown, NJ) and all reagents were analytical grade (Sigma-Aldrich, Inc. FLUKA, St. Louis, MO).
Plasma omeprazole levels were determined by using a HPLC method coupled with mass spectrometry (MS/MS); this method was developed and validated by personnel of Biokinetics in Mexico City, Mexico. The method included the following: 250 μL of plasma, 10 μL of internal standard (pantoprazole sodium, 250 ng/mL) and 750 μL of acetonitrile. These components were vortexed in a 2.0 mL conical tube (Sarstedt AG & Co.) for one minute. The tube was centrifuged at 8000 rpm for five minutes at 20 °C. The supernatant was separated and injected (volume of injection = 1 μL) into the chromatographic system (HPLC, Agilent Technologies, model 1200, Palo Alto, California).
The selected analytical column was a Zorbax® SB-C18, 50 × 4.6-mm internal-diameter column of 1.8-μm particle size (Agilent Technologies). Omeprazole and the internal standard were eluted with a mobile phase consisting of a mixture (75:25 v/v) of 0.1% acid formic and acetonitrile: water (95:5 v/v). A linear flow rate gradient was used and set as follows: 0 to 3.5 min (0.5 mL/min), and 3.6 to 6 min (0.7 mL/min). The column temperature was 25 °C and both analytes were detected by a triple-quadrupole mass spectrometer (Agilent Technologies, model G6410B). The spectrometric (MS/MS) analysis was performed by monitoring the transitions 346.1→198.1 m/z for omeprazole and 384.2→200.1 m/z for the internal standard. The spectrometric conditions were as follows: positive-ionization mode, fragmenter energy (80 V), collision energy (3 V), drying gas (nitrogen at 350 °C), gas flow (12 L/minute) and pressure (45 psi). Typical retention times for omeprazole and the internal standard were 3.3 and 5.4 minutes, respectively. The peak area was measured for calculation of the peak area ratio of omeprazole with respect to the internal standard, and the concentration was calculated.
The analytical method was validated according to Mexican and international guidelines [11,12].
The selectivity of the method was tested by the analysis of blank human plasma samples from six different subjects, blank human (hemolyzed and lipemic) plasma samples, as well as anticoagulants (lithium and sodium heparin), xanthines (caffeine and theobromine), and other drug substances commonly used as analgesics (acetylsalicylic acid, ibuprofen, diclofenac, paracetamol and naproxen). No interferences were observed in the resulting chromatograms.
The calibration curve consisted of the following omeprazole concentrations: 3, 25, 75, 100, 150, 225 and 300 ng/mL. Thus, the range of the method was 3 to 300 ng/mL, with lower limits of quantification (LLOQ) and of detection (LLOD) of 3 and 1 ng/mL, respectively. The method was found to be linear within this range of concentrations with a coefficient of determination of 0.99. The intra-assay %CV and accuracy (relative error) for omeprazole were 0.91% to 2.56% and -5.53% to 0.63%, respectively, while the inter-assay %CV and accuracy were 2.57% to 4.80% and -6.45% to -2.21%. The absolute recovery was above 99.06%.
Omeprazole was found to be stable in plasma after 24 hours at room temperature (25 °C), after three freeze-thaw cycles and after 16 weeks at –75±5 °C.
The validation of the method also included sample dilution to account for omeprazole concentrations beyond the upper bound of the calibration curve’s range.
Quality control (QC) samples were included in every analytical run to verify performance. These QC samples were prepared at three different concentration levels (designated as low (10 ng/mL), medium (125 ng/mL) and high (275 ng/mL) of omeprazole independent of the calibration curve. This method was considered to be suitable by the investigators for evaluating the bioequivalence of omeprazole.
The acceptance criteria for the approval of the analytical runs and the QC samples, as well as the criteria for performing sample reanalysis, were in accordance with Mexican and international guidelines [11,12].
Tolerability was determined using clinical assessment, by monitoring vital signs at baseline, 2 and 6 hours after the drug administration during hospitalization, and at the end of the clinical stage of the study.
The subjects were interviewed (using open-ended questions) by the clinicians concerning the occurrence of AEs during the trial and at the end of the clinical stage of the study. The subjects were asked to spontaneously report any AEs to the clinicians at any time during the study, including during the washout period. The data for all AEs were recorded on a case-report form.
AEs that were life-threatening, led to death, hospitalization, disability, and/or medical intervention to prevent permanent impairment or damage, were considered serious.
Sample size calculation was based on the within-subject variability of omeprazole Cmax with a %CV of 29.3% [13,14]. This calculation was performed considering the following values: 1 - β = 0.8, α = 0.05, %CV = 29.3, and an equivalence range of 80% to 125%, yielded with a sample size of 30 subjects. Thus, we planned to recruit 34 subjects to account for potential dropouts.
Individual plasma concentration–time curves were constructed; the Cmax and Tmax values were directly obtained from these curves, and the area under the plasma concentration-time curve from time baseline to the last measurable concentration (AUC0–t) was calculated by a non-compartmental method using the linear trapezoidal rule. From the terminal log-decay phase, the elimination rate constant (ke) was estimated using linear regression, and the t½ was estimated using the following equation:
t½ = ln2/ke, where ln was defined as the natural logarithm [13].
The extrapolation of AUC from baseline to infinity (AUC0–∞) was calculated as follows:
AUC0–∞ = AUC0–t + Ct/ke,
where Ct was the last measurable plasma concentration.
To assess the bioequivalence between the test and reference formulations, Cmax, AUC0–t and AUC0–∞ were considered as the primary variables. Using log-transformed data for these parameters, ANOVA for a 2 x 2 crossover design, was carried out at the 5% significance level (α = 0.05).
The 90% CIs of the geometric mean ratios (test/reference) of the Cmax, AUC0–t and AUC0–∞ were calculated using log-transformed data. The test and the reference formulations were considered bioequivalent if the 90% CIs of these parameters fell within a predetermined range of 80% to 125%. All pharmacokinetic and statistical analyses were performed using WinNonlin Version 5 (Pharsight, Mountain View, California).
Table 1 shows the demographic characteristics for a total of 34 subjects, who were enrolled in the study.
Two subjects were withdrawn from the study because one subject reported adverse events AE (abdominal pain, nauseas and vomiting) during the washout period and the other subject did not participate in the second period of the clinical stage. Thus, the sample size for the bioequivalence evaluation was reduced from 34 subjects to 32 subjects
Mean plasma concentration-time curves of the two formulations are shown in Figure 1. The results suggest that the two formulations have comparable mean plasma concentration-time curves.
Table 2 shows the pharmacokinetic parameters (Cmax, Tmax, t1/2, AUC0–t and AUC0–∞) for both formulations.
No significant period or sequence effects were detected based on the ANOVA of Cmax, AUC0–t and AUC0–∞ (data not provided).
Table 3 shows the bioequivalence statistics using the log-transformed data for Cmax, AUC0–t and AUC0–∞: geometric means, geometric mean ratios (test/reference), 90% CI, and the intra-subject %CV.
The 90% CIs for omeprazole Cmax, AUC0–t and AUC0–∞ were 84.08% to 104.47%, 87.42% to 108.18%, and 87.94% to 108.15%, respectively. The 90% CIs of the geometric mean ratios of the three parameters fell within the predetermined range of 80% to 125%. It is important to mention that although the data obtained from the two subjects who did not complete the study (only the first period) were not included for the bioequivalence evaluation, a separate statistical analysis, which included the data obtained for these subjects during the first period, revealed that the bioequivalence conclusions were not affected (data not provided). Therefore, these results indicate that the bioequivalence criteria were met.
Ten of the 34 subjects reported a total of 14 AEs. These included two reports of headaches, one that occurred after the administration of the reference formulation and one that occurred after the administration of the test formulation; two reports of nausea that occurred after the administration of the test formulation; two reports of dizziness, one that occurred after the administration of the reference formulation and one that occurred after the administration of the test formulation; one report of abdominal pain after the administration of the test formulation and one case of vomiting after the administration of the test formulation.
Other AEs included a pain of the left wrist hand, a pain of the left forearm, a hematoma on the left forearm, a cheek ecchymosis, a report of fainting and a case of pharyngotonsillitis.
None of the AEs was considered to be serious. Instead, all of the AEs were regarded as mild, and all of the AEs spontaneously resolved under medical surveillance during the clinical stage.
It is important to point out, that one of the 10 subjects who reported AEs experienced abdominal pain, nausea and vomiting during the washout period. Although they AEs were regarded as mild and unrelated to the study formulation, we decided to withdraw this subject from the study (as previously mentioned).
All of the 90% CIs of the geometric mean ratios of the pharmacokinetic parameters (Cmax, AUC0–t and AUC0–∞) were found to be within the predetermined range of bioequivalence (80%-125%). These results indicate that the bioequivalence criteria were met.
Both formulations were well tolerated, as all of the reported AEs were considered to be mild and resolved spontaneously during the clinical stage.
It is interesting to compare the pharmacokinetic data obtained in the current study to those data obtained by Poo, et al. in their bioequivalence study for the enteric-coated formulations of omeprazole 20 mg in a Mexican population [14]. For the current study, the reference-formulation means (± standard deviation) of the Cmax, Tmax and AUC0–∞ values were 760.33 (±355.36) ng/mL, 0.54(±0.39) hours, and 993.25 (±680.39) ng•h/mL, respectively, whereas the corresponding values obtained for the reference (enteric-coated) formulation were 490 (±260) ng/mL, 1.9 (±0.8 hours), and 1000 (780) ng•h/mL, respectively. This suggests that there are differences in the Cmax and Tmax values between the combination of omeprazole with sodium bicarbonate and the enteric-coated formulation, which is consistent with the reported faster absorption of omeprazole, with shorter times to peak plasma concentrations for the combination of omeprazole with sodium bicarbonate, compared to the enteric-coated formulations [5].
As with any clinical trial, particularly bioavailability studies, the current study had some limitations that should be considered. First, this was a single-blind study, so it might not objectively address the effectiveness and safety profiles of the tested formulations.
The data were obtained from healthy adult subjects within a specific age range, who were administered a single dose of the formulation, in accordance with regulatory requirements (COFEPRIS). The pharmacokinetic parameters of omeprazole in combination with sodium bicarbonate might differ in target populations. The pharmacokinetics of this combination might also differ among patients in different ethnic groups and with different genotypes [15]. Thus, the results of this study might not be generalizable to a target population.
In addition, an evaluation of the effect of food on the bioavailability of omeprazole in combination with sodium bicarbonate was not considered because this combination should be administered on an empty stomach one hour prior to a meal [7].
Further studies are needed to compare the test formulation with the reference formulation in Mexican patient groups. The results of this study might serve as a reference for future controlled studies of omeprazole in combination with sodium bicarbonate in a Hispanic population.
In this study of healthy, fasting, Mexican adult subjects, who received a single dose of either the test or the reference formulation, we concluded that the test formulation of omeprazole 20 mg in combination with 1100 mg of sodium bicarbonate met the Mexican regulatory requirements to assume bioequivalence, based on the rate and extent of absorption. Both formulations were well tolerated.
This research and its publication were supported by Laboratorios Liomont, S.A. de C.V., Mexico City, Mexico. The authors have indicated that they have no conflicts of interest regarding the content of the article.

![]()

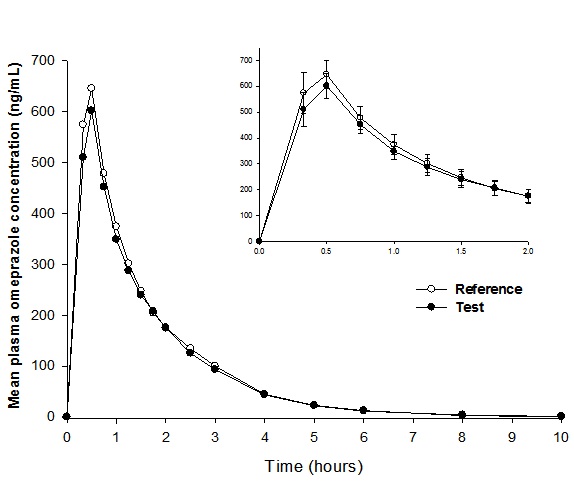 |
| Figure 1: Mean plasma concentration-time curves after a single-dose administration of a test (trademark: Inhibitron Twit®, Laboratorios Liomont, S. A. de C. V.) and a reference (trademark: Trinsica® 20, GlaxoSmithKline Mexico, S. A. de C. V.) oral capsule, containing omeprazole 20 mg in combination with 1100 mg of sodium bicarbonate in healthy Mexican adult subjects (n = 32). Inset: mean (±SE) concentrations over the first 2 hours after administration |
Characteristic |
Values |
|---|---|
Total No. of subjects (female/male) |
34 (10/24) |
Age, mean (SD), range, years |
37 (9), 21-50 |
Weight, mean (SD), range, kg |
65.34 (6.05), 55.30-83.30 |
Height, mean (SD), range, m |
1.66 (0.09), 1.51-1.95 |
BMI , mean (SD), range, kg/m2 |
23.74 (1.50), 20.31-25.91 |
BMI = Body mass index |
|
Parameter |
Reference† |
Test* |
|---|---|---|
Cmax, μg/ml |
760.33 (355.36) |
703.75 (283.11) |
AUC0–t, μg·h /ml |
968.52 (669.86) |
916.53 (641.71) |
AUC0–∞, μg·h/ml |
993.25 (680.39) |
943.02 (656.88) |
Tmax, h |
0.54 (0.39) |
0.49 (0.26) |
t1/2, h |
0.86 (0.31) |
0.86 (0.33) |
| Cmax = Maximum plasma drug concentration AUC0–t = AUC from time 0 (baseline) to the last measurable concentration AUC0–∞ = AUC from baseline extrapolated to infinity *Trademark: Garbican® (Laboratorios Liomont, S. A. de C. V., Mexico City, Mexico) †Trademark: Lyrica® (Pfizer, S. A. de C. V., Mexico City, Mexico) Table 2: Pharmacokinetic parameters of a reference and a test formulation of omeprazole after a single-dose administration of omeprazole 20 mg in combination with sodium bicarbonate 1100 mg in healthy Mexican adult subjects (n = 32). Values are mean (SD) |
||
Parameter |
Geometric Means Test/Reference |
Geometric Mean Ratio (%) |
90% CI |
Intra-subject %CV |
|---|---|---|---|---|
Cmax, ng/mL |
643.71/686.82 |
93.72 |
84.08, 104.47 |
26.01 |
AUC0–t, ng•h/mL |
746.96/768.10 |
97.25 |
87.42, 108.18 |
25.52 |
AUC0–∞, ng•h/mL |
772.61/792.20 |
97.53 |
87.94, 108.15 |
24.74 |
| Cmax = Maximum plasma drug concentration AUC0–t = AUC from time 0 (baseline) to the last measurable concentration AUC0–∞ = AUC from baseline extrapolated to infinity Table 3: Geometric means, geometric mean ratios, 90% CIs and the intra-subject %CV of the pharmacokinetic parameters determined for omeprazole after a single-dose administration of omeprazole 20 mg in combination with sodium bicarbonate 11000 mg in healthy Mexican adult subjects |
||||




































