Open Access
Research Article
Max Screen >>
ISSN: 2454-499X
Copyright: © 2017 Monteiro Cunha JP. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Related article at Pubmed, Google Scholar
The distribution of different human immunodeficiency virus type 1 (HIV-1) genotypes and the prevalence of transmitted drug resistance (TDR) mutations vary greatly across different Brazilian regions. This study aimed to describe the HIV-1 molecular diversity and TDR prevalence among treatment-naïve HIV-1 infected individuals in an urban area of Northeastern Brazil. DNA samples from 97 infected individuals were obtained and pol sequences were generated by Polimerase Chain Reaction (PCR) and direct sequencing. Bioinformatics tools were used to identify the presence of associated mutations with drug resistance, to reconstruct the phylogeny and to detect recombination. The median age and estimated time of HIV-1 diagnosis were 34 years and 12 months, respectively and 61% of the patients were male. The main exposure category was sexual (87.5%) and the median TCD4+ lymphocyte counts and viral load were 438 cells/mm3 and 4.4 log10, respectively. The overall TDR prevalence was 5.1% while within each drug class (Non-nucleoside reverse-transcriptase inhibitors - NNRTIs, Nucleoside analog reverse-transcriptase inhibitors - NRTIs and Protease inhibitors - PIs) this rate was 2.1% individually. These TDR mutations confer intermediate to high-level resistance to different drugs that are used as first-line treatment options, which reinforce the importance of genotype testing prior to treatment initia-tion. Phylogenetic analysis showed that 73.2% of the samples were subtype B, 14.4% were subtype F, 4.1% were subtype C and 8.3% were BF recombinants. Our data indicate that the genotypic profile of HIV-1 strains circulating in Bahia is diversified and show an increase of subtype C prevalence in Northeast Brazil.
Keywords: Genotyping; Mutation; Antiretroviral; Subtypes; HIV; Resistance; Prevalence
Despite the undeniable results coming from Highly Active Antiretroviral Therapy (HAART), the selection and dissemination of Human Immunodeficiency Virus (HIV) resistant strains contribute negatively to life quality and increase the risk, up to 13 times, of failure in obtaining undetectable viral loads. The United States and European countries guidelines suggest that genotyping test should be performed prior to the introduction of antiretroviral drugs in regions with transmitted drug resistance (TDR) prevalence higher than 5%. This recommendation, although grounded in cost-effectiveness studies and in the impactful reduction of first scheme failure risk, has not been adopted yet by the Brazilian guidelines.
In 2009, the World Health Organization (WHO) recommended the use of an epidemiological monitoring list with the purpose of methodological standardization among TDR prevalence reports. The surveillance drug resistance mutations (SDRMs) list is based on mutations that alter sensitivity to one or more drugs and are com-mon to all HIV-1 subtypes, as long as they are not polymorphisms or very rare [1,2].
The characterization of the molecular epidemiology through phylogenetic analyzes contributes to a better understanding of the dynamic processes of HIV/AIDS (Acquired immunodeficiency syndrome) spread and to the identification of regional patterns of virus dissemination, therefore contributing to the development of specific control measures for each locality. In this context, the Brazilian territory is divided into five geographic regions that present different patterns of HIV-1 subtype distribution. Besides, these prevalence patterns seem to be constantly changing over time. In different regions of the country, the HIV epidemic previously characterized by subtype B predominance has been associated with subtype C and recombinant forms growth. In Northeastern Brazil, subtype B predominates with co-circulation of subtype F, BF, and BC recombinant forms [3,4]. This study aimed to describe the molecular epidemiology of HIV-1 and to estimate the TDR prevalence among treatment naïve infected individuals in Salvador, the capital of the state of Bahia, located in the northeastern region of Brazil. The results described here may assist in decision-making in health policies, such as the recommendation for the performance of genotyping in all patients who initiate therapy and therapeutic orientation of the initial scheme with greater chances of success in viral suppression.
The present study included a convenience sample consisting of 97 HIV-1 infected patients that were 18 years and treatment-naïve. These individuals were regularly followed at the Professor Edgard Santos University Hos-pital, or at the Specialized Center for Diagnosis, Care and Research in AIDS, STDs and Viral Hepatitis (CEDAP) and were randomly invited to participate in the study during their routine medical visit. They signed a term of informed consent to participate in this study, which was approved by the Oswaldo Cruz Foundation (FIOCRUZ) Institutional Review Board. Blood samples were collected during 2012 and sent to the Laboratory of Hematology, Genetics and Computational Biology/CPqGM/FIOCRUZ for processing. Clinical and epidemiological data were obtained from patient medical records and interviews.
Genomic DNA was extracted from whole blood using a QIAamp DNA Blood Mini Kit (QIAGEN, Valencia, CA) and amplified by nested Polymerase Chain Reaction (PCR) using POL1 (GGGAGTGGGGG-GACCCGGCCATAA) and INBO1-R (CCACTCAGGAATCCAGGTGG) primers in the first round, and K1 (CAGAGCCAACAGCCCCACCA) and K2 (TTTCCCCACTAACTTCTGTATGTCATTGAC) primers in the second round. Cycling conditions were as follows: one cycle of denaturing at 94 °C for 5 min; 35 cycles of denaturing at 94 °C for 45s, annealing at 55 °C for 45s and primer extension at 72 °C for 2.5 min with a final extension at 72 °C for 7 min. The amplified fragment corresponded to nucleotide positions 2358 to 3161 in the HXB2 reference genome (amino acid positions 36 to 99 in the Protease (PR) region and 1 to 204 in the Reverse Transcriptase (RT) region).
All PCR products were purified using Qiagen columns (QIAGEN) and sequenced in an ABI 3100 Genetic Ana-lyzer (Applied Biosystems, Foster City, CA) using the Big Dye Terminator kit (Applied Biosystems) and DP10 (CAACTCCCTCTCAGAAGCAGGAGCCG),DP16 (CCTCAAATCACTCTTTGGCAAC) F1 (GTT-GACTCAGATTGGTTGGAC) and F2 (GTATGTCATTGACAGTCCAGC) primers. DNA sequences were assembled using Geneious 6.1.6 software and checked for contamination using the HIV Quality Analysis Pipe-line Tool ( https://www.sanbi.ac.za ). All data are available in the GenBank database ( https://www.ncbi.nlm.nih.gov/genbank/ ). under the Accession numbers KT950963-KT951059.
The obtained sequences were aligned with reference sequences from Genebank using the Clustal X multiple sequence alignment program [5]. The alignment was manually edited using the Se-Al program ( https://tree.bio.ed.ac.uk/software/seal/ ) and submitted to the jModelTest [6] program for evolutionary model selection. Neighbor-joining (NJ) and maximum likelihood (ML) trees were constructed using PAUP* 4.0b10 software [7]. Node reliability was assessed by bootstrap analysis (1000 replicates) and the likelihood ratio test was used to calculate statistical support for the branches. Trees were interpreted using the FigTree program version 1.3.1 ( https://tree.bio.ed.ac.uk/software ). Recombination was identified by the bootscanning method implemented in SimPlot software [8]. Crossover points were determined by visual inspection of the alignment using GeneDoc software [9] and confirmed by the phylogenetic reconstruction of trees using the individual crossover points. The identified BF recombinant sequences were aligned with CRF reference sequences ob-tained from the Los Alamos database ( https://www.hiv.lanl.gov/content/index ) and analyzed by phylogenetic reconstruction as described above.
The prevalence of drug-resistant mutations was determined using the SDRMs list. The Stanford HIV Drug Re-sistance Database algorithm was also used to identify mutations known to be associated with reduced phenotypic drug susceptibility or with reduced virological response in clinical practice ( https://hivdb.stanford.edu/ ).
Epidemiological, clinical and genetic data were analyzed using SPSS version 13.0 (Chicago, IL, USA).
The clinical, demographic and epidemiological characteristics of the 97 HIV-1 infected patients are shown in Table 1. Among these, 62 (63.91%) were male, age ranged from 19 to 62 years and the estimated time of posi-tive HIV-1 diagnosis ranged from 0 to 252 months. The main exposure factor was sexual contact, reported by 85 (87.6%) individuals, followed by sharps injuries (3%) and intravenous drug use (IDU) (1%). Eight patients (8.2%) did not provide information regarding mode of transmission. The median TCD4+ lymphocyte and viral load counts were 438 cells/mm3 and 4.4 log10 respectively.
Phylogenetic analysis revealed that 71 samples (73.2%) belonged to subtype B, 14 (14.4%) were subtype F, four (4.1%) were subtype C, and eight (8.3%) were BF recombinants that grouped into three distinct patterns of recombination. One of these groups consisted of samples BA07PV, BA11PIR, BA44VAL, BA80BRT and BA69PNB, which formed a monophyletic cluster (Figure 1), exhibiting the same recombination pattern as other BF recombinant samples previously identified in Bahia (Figures 2a and 2b) [3,4]. These five sequences also clustered within a larger clade along with CRF28 and CRF29 genome sequences, which share the same recom-bination profile in this genomic region. Another BF recombination pattern was identified in samples BA06PLT and BA112LIB, which similarly grouped very closely to two other BF sequences previously identified in Bahia (Figure 1), again sharing the same recombination pattern (Figures 2c and 2d) [4]. In addition, these four se-quences formed a larger clade along with other CRF12 genome sequences. The third BF recombination pattern was observed in one sample (BA111ITP), which grouped together with another previously identified sequence from Bahia (BAS042) within a larger cluster of CRF39 genome sequences [10]. The close phylogenetic rela-tionship between these two sequences from Bahia was confirmed by SimPlot analysis (Figure 2e and 2f).
SDRMs analysis revealed that four patients (4.1%) presented TDR to at least one class of antiretroviral drugs SDRMs (Table 2). Two (2.1%) of these presented TDR to NNRTIs, one (1%) to NRTIs and two (2.1%) to PIs. Their median age, estimated time of diagnosis (time elapsed between the date of diagnosis and the date of sample collection), CD4+ lymphocyte count, and viral load were 39.5 years, 17.5 months, 523 cells/mm3 and 4.9 log10, respectively. These patients’ viral strains were classified as subtype B (n=3) and as CRF 39_BF-like recombinant (n=1). One of these (BA91CN) presented two mutations associated with resistance to PIs (M46I and V82T), while another (BA114NVB) presented two mutations associated with resistance to two different antiretroviral classes, NRTIs (M41L) and PIs (L90M).
According to the Stanford HIVdb algorithm, 25 (25.8%) samples harbored at least one mutation associated with any level of antiretroviral resistance (Table 3), while 12 (12.3%) samples had mutations associated with resistance to NNRTIs, 12 (12.3%) to PIs and 2 (2.1%) to NRTIs .
The most prevalent mutational profiles associated with each antiretroviral class were: ten (10.3%) A71T/V/I and one T74S, M46I, V82T, L90M and Q58E (1.0% each) associated with PIs; six (6.2%) E138A, two (2.1%) V90I and one K103N, V179D, V108I, K101H and G190A (1.0% each) associated with NNRTIs; one (1.0%) M41L and one (1.0%) K65N associated with NRTIs.
The present study evaluated the prevalence of transmitted drug resistance mutations in a convenience sample from Salvador, the capital of the state of Bahia, located in the northeastern region of Brazil, using the SDRMs list recommended by the World Health Organization (WHO), last updated in 2009 [2]. TDR prevalence was estimated at 4.1% in this drug-naïve study sample, which is considered low by the WHO. This finding is con-sistent with a previous study conducted in the city of Salvador in 2009, in which a TDR prevalence below 5% was reported [13]. Furthermore, the individuals enrolled in the previous study presented similar mean TCD4+ (577 cells/mm3) counts and viral loads (4.58 log10) to those observed herein (mean TCD4+ of 454 cells/mm3 and mean viral load of 4.3 log10). However, the TDR prevalence reported here is markedly lower than that described by De Moraes et al (17%), whose samples were collected in the same year as ours (2012) [12]. It is interesting to note that these authors included 47 individuals from Salvador who were also about to initiate antiretroviral treatment, but whose mean TCD4+ lymphocyte counts (227 cells/mm3) were lower, while viral loads (5.2 log10) were higher, in comparison to those observed in the present study.
The low drug resistance prevalence found in the present study might also be related to the genomic origin of the analyzed sequences. Significantly fewer mutations have been found in proviral DNA compared to historic viral RNA genotypes showing that some resistance mutations are not archived or are not detectable in the latent reservoir (Wirden et al. 2011).
Of note is that the WHO drug resistance mutation list was most recently updated in 2009, whereas the Stanford algorithm receives updates more frequently (last updated in 2016). Importantly, the 2009 WHO guidelines do not recognize the K65N mutation as being associated with drug resistance. The Stanford algorithm associates a virus harboring K65N, a non-polymorphic mutation associated with reduced susceptibility to several NRTIs [13,14], with intermediate-level resistance to Tenofovir (TDF), Lamivudine/Emtricitabine (3TC/FTC), Abacavir (ABC), Stavudine (D4T), and Didanosine (ddI). K65N is a rare mutation, showing a < 1% frequency of K65N among drug naïve individuals ( https://hivdb.stanford.edu/DR/ ). This mutation was linked to a dramatic decline in HIV replication capacity and presents similar but less pronounced effects on NRTI susceptibility than K65R [14,15]. Considering the fact that the K65N mutation was identified by the Stanford algorithm analysis in one of our included samples, this shifts the overall TDR prevalence to 5.1%, which then becomes classified as an intermediate level of primary drug resistance. Accordingly, the TDR associated with NRTIs in light of the K65N mutation increases to 2.1% in this population under the Stanford algorithm. In addition, the sequence amplifications analyzed in the present report ranged from amino acid positions 33 to 99 in PR and from 1 to 204 in RT, which could result in the underestimation of TDR prevalence due to a lack of detection of mutations associated with resistance occurring either before or after the targeted positions. For example, it was not possible to identify the prevalence of mutations at positions 210 and 215, thus probably underestimating resistance to NRTI and the possible impact of the M41L mutation.
Upon considering the potential impact of the TDR mutations detected herein, it becomes evident that the effec-tiveness of first-line antiretroviral therapies could be compromised. The mutations found in our study population, e.g. G190A, K103N, L90M, M46I and V82T, confer changes in drug sensitivity and have been associated with intermediate to high levels of resistance to first-line drugs for early therapy, such as Efavirenz, Nevirapine, Atazanavir/Ritonavir and Lopinavir/Ritonavir. The mutations detected herein also confer, to a lesser degree, changes in sensitivity to Etravirine and Tipranavir/Ritonavir, which are reserved for patients exhibiting advanced levels of treatment failure in response to antiretroviral drugs ( https://hivdb.stanford.edu/DR/ ).
This study also investigated the molecular epidemiology of the HIV-1 subtypes circulating in the state of Bahia in 2012. Subtype B (73.2%) was most prevalent, followed by subtype F (14.4%), subtype C (4.1%) and BF recombinant forms (8.3%). Compared with the subtype prevalence rates reported by Santos and colleagues (2011) in Salvador, our results indicate a decrease in subtype B (77.2%) and in BF recombinants (21.0%) and an increase in subtype F (1.8%) prevalence [4]. The prevalence of subtype C was found to be significantly higher (p < 0.05) than that reported in 2011 (1.7%) [3], which points to the establishment and expansion of subtype C in the Brazilian Northeast. Among the samples classified as BF recombinant, five HIV-1 isolates shared the same recombination pattern with 18 BF sequences previously reported in Bahia (Figure 1). Considering the viral isolates identified in the state of Bahia to date, there appears to be a predominant BF recombinant form circulating among the other recombinant types detected here, as well as in previous reports [4,16]. Taking into account that the point of recombination of these variants occurred in the same region as the point of recombination of CRFs 28 and 29, together with the clustering profile of the BF recombinant samples seen in the phylogenetic tree, it is possible that these viral isolates may either belong to these forms of HIV-1, or they may potentially represent a novel HIV-1 CRF, as proposed in previous reports [4,16]. This is indeed possible, because despite the fact that CRF 28 and 29 have the same common point of recombination in this region of the genome, they are nonetheless classified as different CRFs due to differences in other recombination points. In order to further clarify this finding, it would be necessary to sequence the full genome of these recombinant samples. In addition, two other recombinants identified here clustered with other samples from Bahia and with CRF12 sequences. Similarly, these viral isolates may actually be representative of the introduction and consequent spread of CRF12 in the region, or they may represent a novel HIV-1 CRF. Again, to elucidate this, the complete sequencing of these genomes is required. Finally, another sequence (BA111ITP) that clustered with CRF39 genome sequences was previously identified in the southeastern region of Brazil, which might indicate the introduction of this variant in the Northeast.
In conclusion, the transmitted resistance rate found in the present study sample from Salvador, Bahia-Brazil, is classified as intermediate according to the Stanford algorithm (5.1%), but low according to the WHO (4.1%). The TDR mutations found in this study population confer intermediate to high levels of resistance to several antiretroviral drugs used as first-line treatment options. Is it clear that the adoption of genotype testing prior to the initiation of HAART can increase the chances of success with respect to this Brazilian population. Moreover, an accumulating body of evidence supports the hypothesis that a novel BF recombinant form could be circulating in Bahia, and that the subtype C epidemic is increasing in this region of Brazil.
This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).

![]()

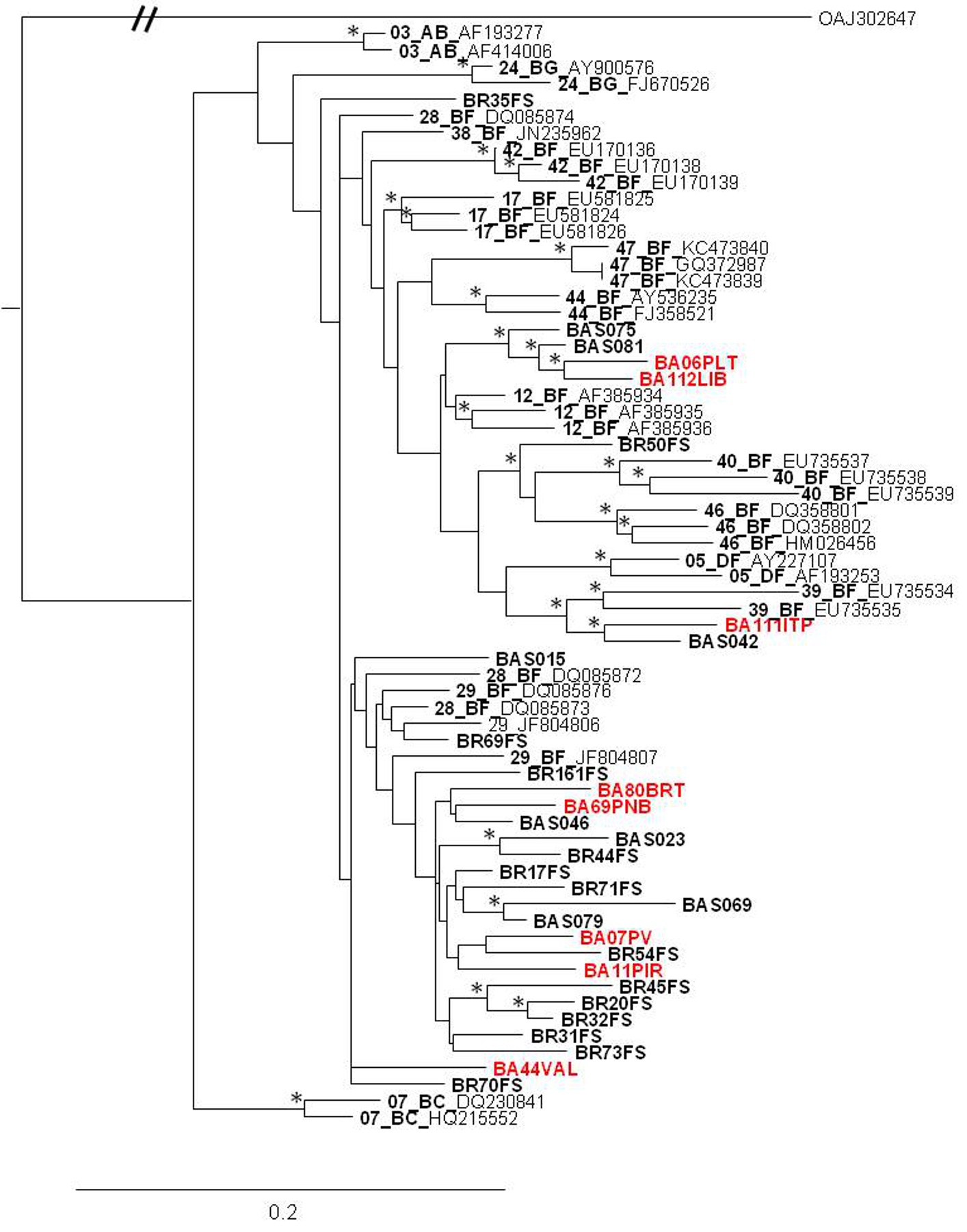 |
| Figure 1: Phylogenetic relationship of the recombinant viruses identified in this study and other HIV-1 recombinant viruses based on pol sequences. Samples generated here are showed in red while BF recombinants previously identified in Bahia state are showed in bold. A Group O sequence was used as outgroup. Trees were constructed based on the Maximum Likelihood method. Branch lengths were estimated with the best fitting nucleotide substitution model according to a hierarchical likelihood ratio test, and were drawn to scale with the bar at the bottom indicating 0.2 nucleotide substitutions per site. The asterisk (*) along the branches represents significant statistical significance for the clade subtending that branch (bootstrap value > 70%) |
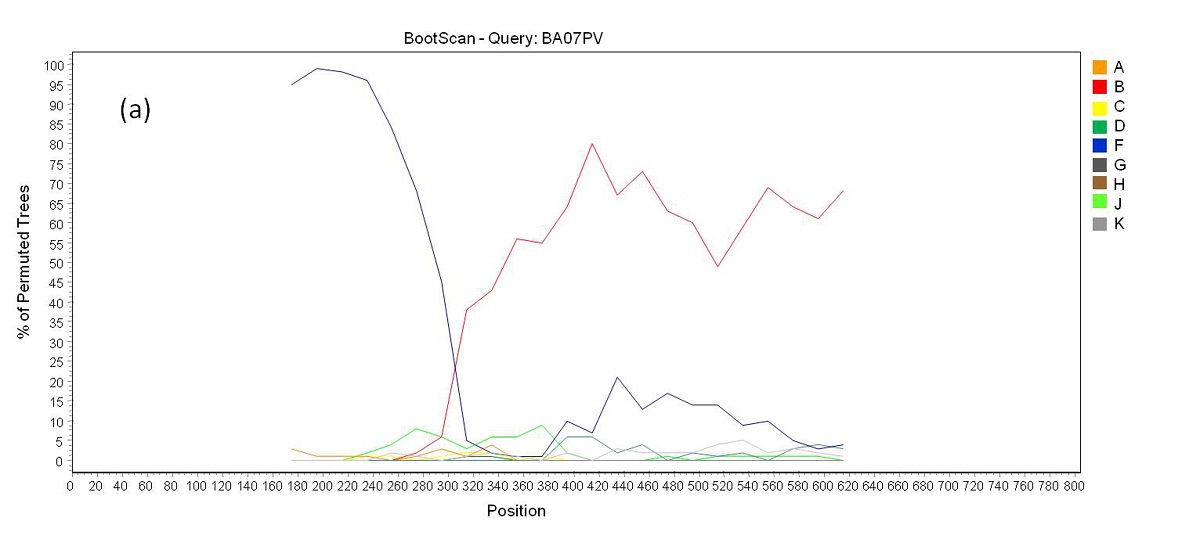 |
| Figure 2a: Bootscan analysis of BF recombinants. (a) Recombination pattern between subtypes B an F inside the pol gene of samples BA07PV, BA11PIR, BA44VAL, BA80BRT and BA69PNB |
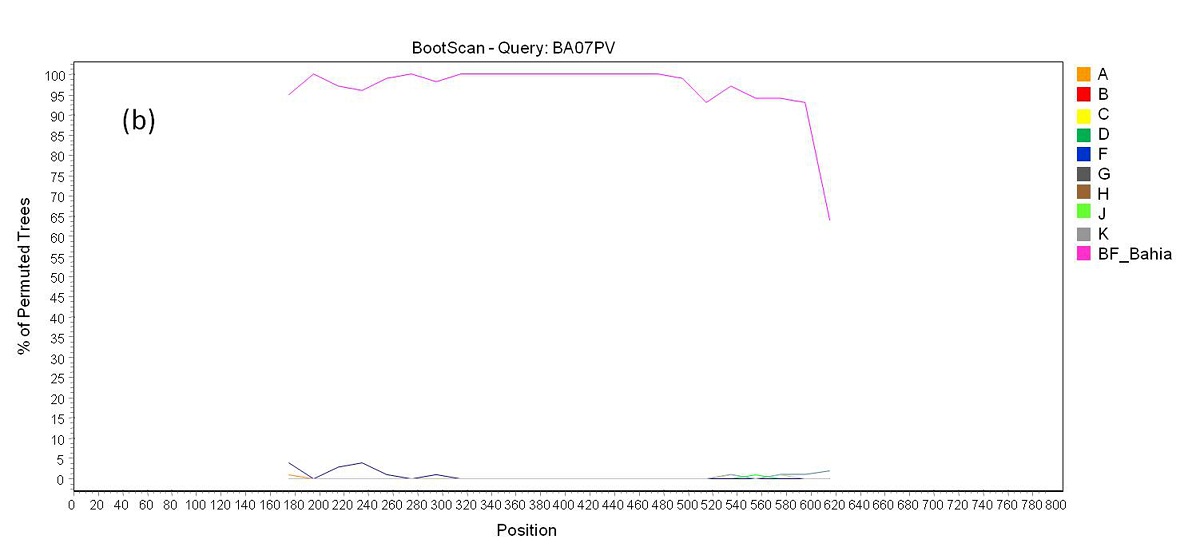 |
| Figure 2b: Bootscan analysis of BF recombinants. (b) a reference group formed by BF samples from Bahia state previously reported were included in the analysis. Each sample above showed a closer phylogenetic relation to this reference group (pink line) than to the pure subtypes B and F reference groups through all the sequence length |
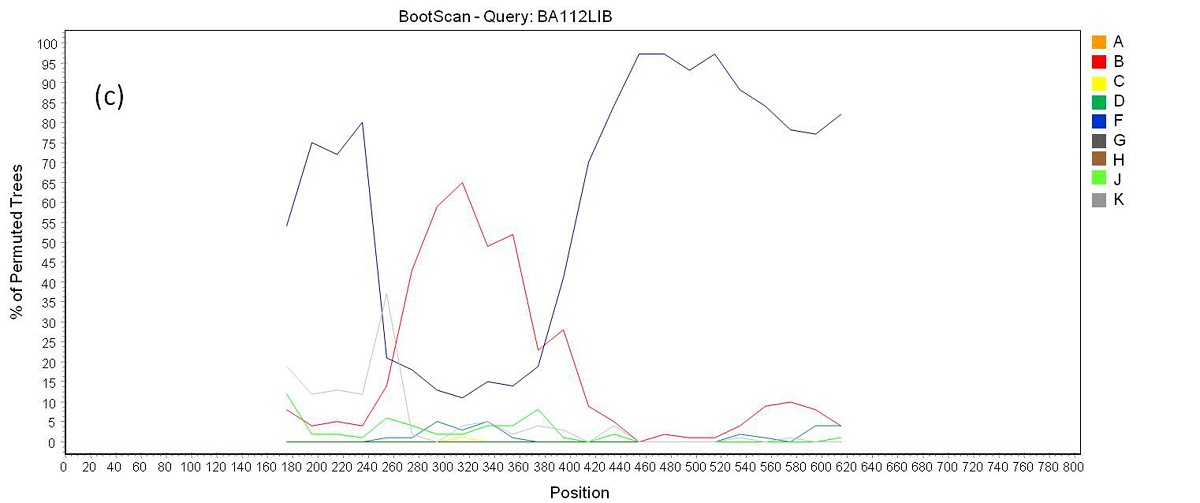 |
| Figure 2c: Bootscan analysis of BF recombinants. (c) Recombination pattern between subtypes B an F inside the pol gene of samples BA06PLT and BA112LIB |
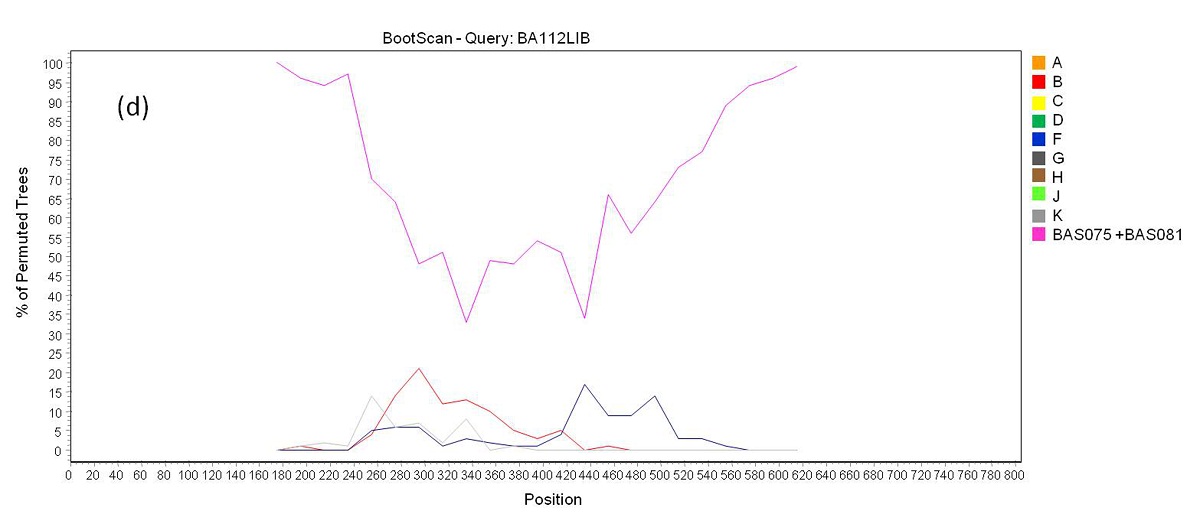 |
| Figure 2d: Bootscan analysis of BF recombinants. (d) a group formed by sequences BAS075 and BAS081 from Bahia identified in a previous study were included in the analysis. Samples BA06PLT and BA112LIB showed a closer phylogenetic relation to this reference group (pink line) than to the pure subtypes B and F reference groups through all the sequence length |
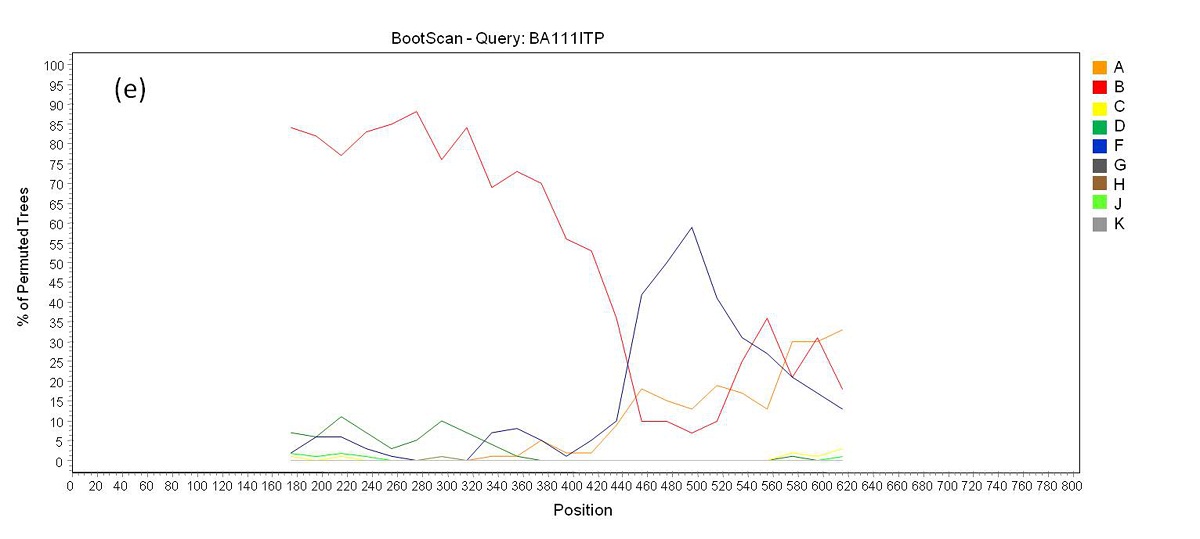 |
| Figure 2e: Bootscan analysis of BF recombinants. (e) Recombination pattern between subtypes B an F inside the pol gene of sample BA111ITP |
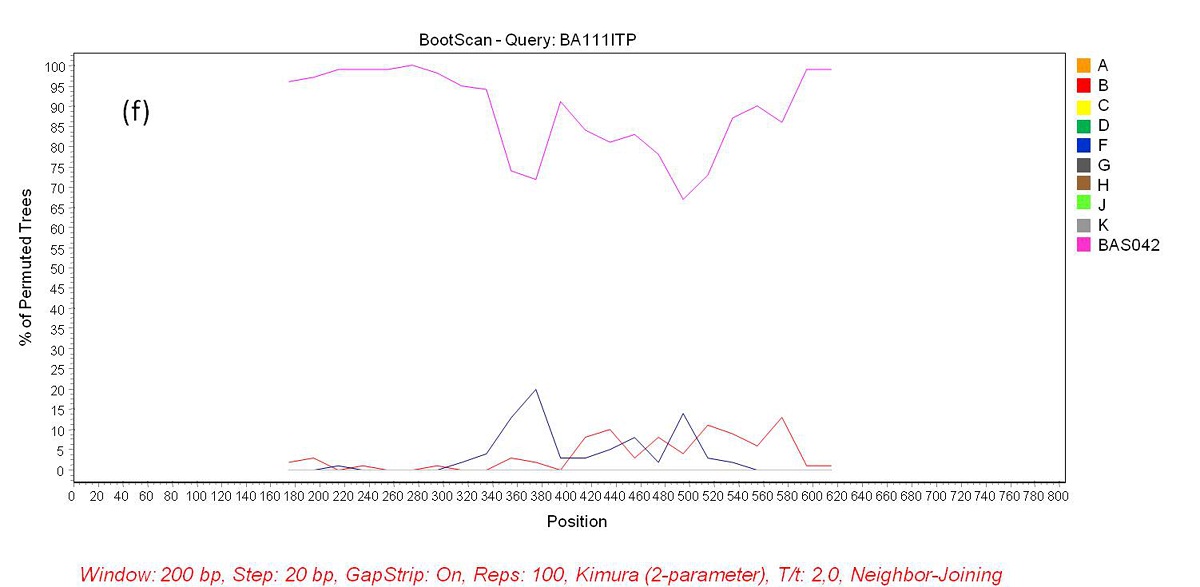 |
| Figure 2f: Bootscan analysis of BF recombinants. (f) Sequence BAS042 identified in a previous study was included in the analysis as a reference group. Sample BA111ITP showed a closer phylogenetic relation to this reference group (pink line) than to the pure subtypes B and F reference groups through all the sequence length |
Characteristics |
n=97 |
|---|---|
Age (years) |
|
Minimum |
19 |
Median |
34 |
Mean |
35.3 |
Maxim |
62 |
Gender (%) |
|
Male |
62 (63.91%) |
Female |
35 (36.09%) |
Estimated time of HIV-1 diagnosis (months) |
|
Minimum |
0 |
Median |
12 |
Mean |
30 |
Maxim |
252 |
Exposure Category |
|
Heterosexual |
48 (49.5%) |
MSM |
36 (37%) |
Bisexual |
1 (1%) |
IDU |
1 (1%) |
Accident with needlestick |
3 (3%) |
Others |
8 (8.5%) |
T CD4+ lymphocytes count (cell/mm3) |
|
Minimum |
09 |
Median |
438 |
Mean |
454 |
Maxim |
1084 |
Viral load (log10)* |
|
Minimum |
2.0 |
Median |
4.4 |
Mean |
4.3 |
Maxim |
5.7 |
MSM: men who have sex with men |
|
Sequence |
Subtype |
TDR |
TDR |
TDR |
|---|---|---|---|---|
BA25CSA |
B |
K103N |
||
BA91CN |
B |
M46I / V82T |
||
BA111ITP |
CRF39_BF |
G190A |
||
BA114NVB |
B |
M41L |
L90M |
|
| Table 2: Mutations associated with primary resistance in the polgene (PR and TR) identified by The World Health Organization 2009 List of Mutations for Surveillance of Transmitted Drug Resistant HIV Strains | ||||
Sequence |
Subtype |
TDR NRTI |
TDR NNRTI |
TDR PI |
Level of Resistance |
|||
|---|---|---|---|---|---|---|---|---|
Potential |
Low |
Intermediate |
High |
|||||
BA03LIB |
B |
E138A |
ETR |
RPV |
||||
BA06PLT |
BF |
A71T (minor) |
||||||
BA08CAB |
B |
E138A |
ETR |
RPV |
||||
BA18CAJ |
B |
V90I |
||||||
BA23DDA |
B |
E138A |
ETR |
RPV |
||||
BA25CAZ |
B |
K103N* |
EFV, NVP |
|||||
BA33PLT |
B |
A71T (minor) |
||||||
BA38PNB |
B |
A71T (minor) |
||||||
BA43PIT |
B |
A71T (minor) |
||||||
BA52MUS |
C |
E138A |
ETR |
RPV |
||||
BA58ITP |
C |
T74S ( minor) |
NFV |
|||||
BA62SUS |
C |
V90I |
||||||
BA65CPF |
B |
A71T (minor) |
||||||
BA73RIV |
B |
A71V (minor) |
||||||
BA75SAM |
B |
E138A |
ETR |
RPV |
||||
BA77PER |
B |
V179D* |
EFV,ETR,NVP, RPV |
|||||
BA78POR |
B |
V108I |
EFV |
NVP |
||||
BA91CAN |
B |
M46I*, V82T* ( major) A71T ( minor) |
SQV/r |
ATV/r, FPV/r, IDV/r, LPV/r, TPV/r |
NFV |
|||
BA97PER |
B |
A71V ( minor) |
||||||
BA105PIT |
B |
E138A |
ETR |
RPV |
||||
BA111ITP |
BF |
K101H G190A* |
ETR, RPV |
EFV, NVP |
||||
BA114NVB |
B |
M41L* |
A71I (minor) L90M* (major) |
ABC, DDI, TDF |
AZT, D4T FPV/r, LPV/r |
ATV/r, IDV/r, SQV/r |
NFV |
|
BA115ITP |
B |
K65N |
3TC, ABC, D4T, DDI, FTC, TDF |
|||||
BA119MDD |
B |
Q58E (minor) |
TPV/r |
|||||
BA121CNT |
B |
A71V ( minor) |
||||||
*Mutations also present in the WHO SDRMs list |
||||||||
• ABC: Abacavir |
• TDF: Tenofovir |
|||||||
• ATV: Atazanavir |
• 3TC: lamivudine |
|||||||
• AZT: Zidovudine |
• FTC: emtricitabine |
|||||||
• D4T: Estavudine |
• TPV/r: Tipranavir/r |
|||||||
• DDI: Didanosine |
• PI: Protease inhibitors |
|||||||
• EFV: Efavirenz |
• NRTI: Nucleoside analog reverse-transcriptase inhibitors |
|||||||
• ETR: Etravirine |
• NNRTI: Non-nucleoside reverse-transcriptase inhibitors |
|||||||
• RPV: Rilpivirine |
• TDR: Transmited Drug Resistance |
|||||||
• NFV: Nelfinavir |
||||||||
• FPV/r: Fosamprenavir/r |
||||||||
• IDV/r: indinavir/r |
||||||||
• NFV: nelfinavir |
||||||||
• LPV/r: Lopinavir/r |
||||||||
• NVP: Nevirapine |
||||||||
• SQV/r: Saquinavir/r |
||||||||
| Table 3: Description of the mutations associated with antiretroviral primary resistance found in the analyzed pol region identified by the Stanford Algorithm | ||||||||




































